The Role of Ataxia Telangiectasia Mutant and Rad3-Related DNA Damage Response in Pathogenesis of Human Papillomavirus



Abstract
1. Introduction
2. Epidemiology and Current Prophylactic Strategies against HPV Infection
2.1. Epidemiological Characteristics
2.2. Cervical Cancer Screening
2.3. The Pros and Cons of the HPV Vaccines against Cancer
3. HPV Life Cycle
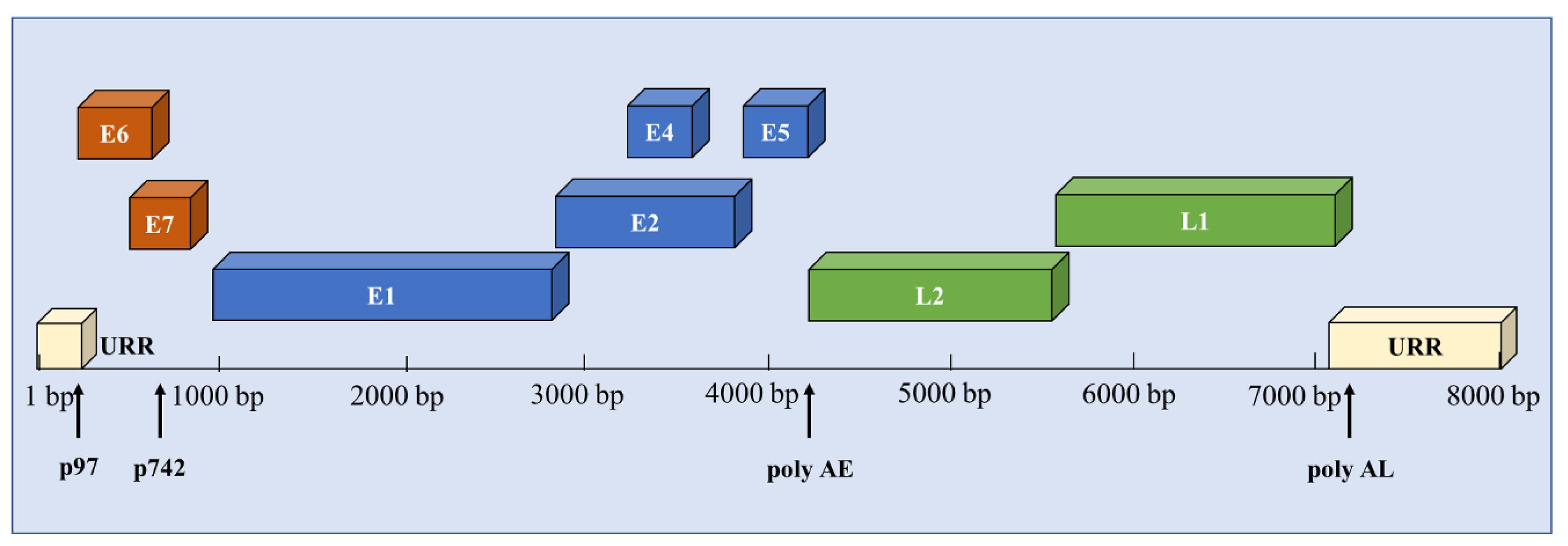
3.1. The Structure of HPV and the Function of Viral Proteins
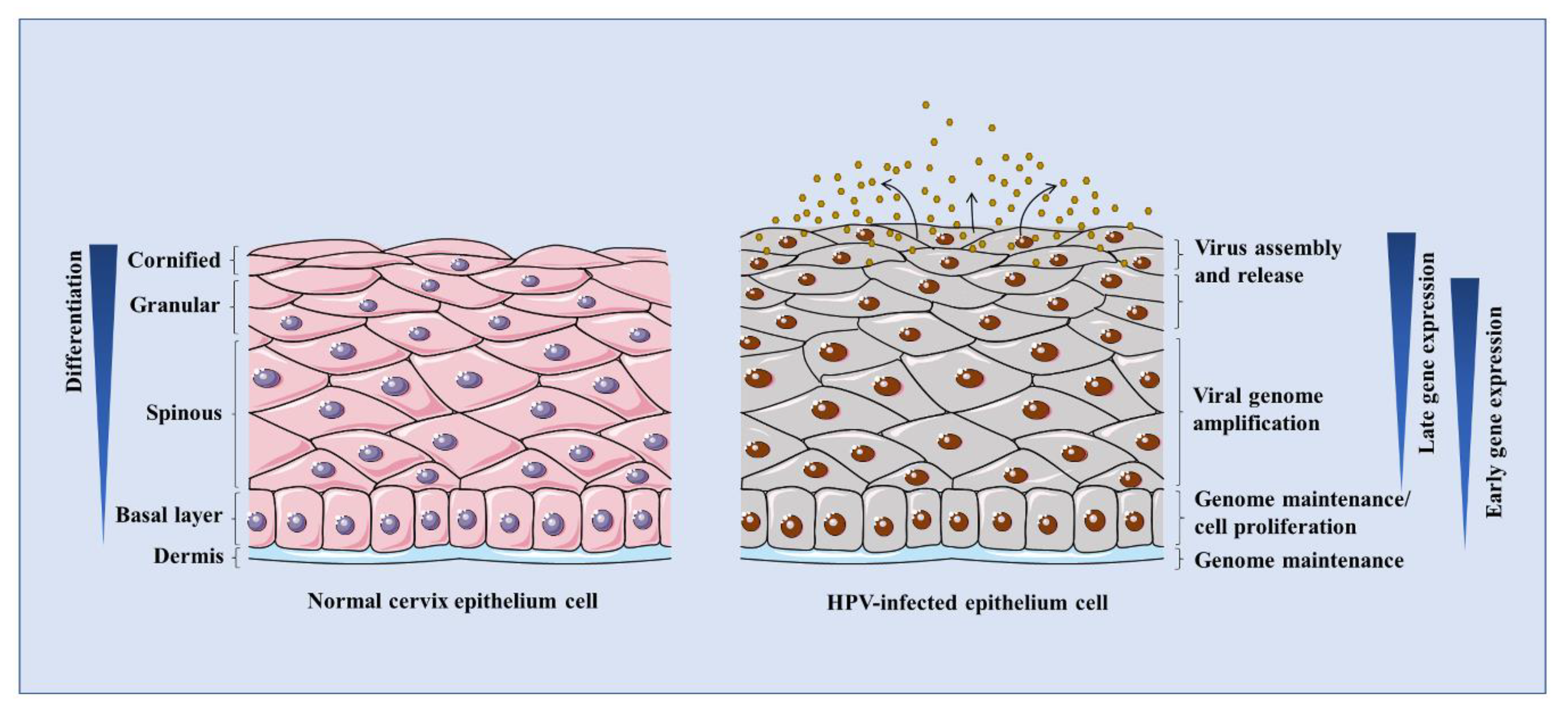
3.2. The Life Cycle of HPV
3.3. Mechanisms of HPV Life Cycle
4. The Role of ATR DDR in the Molecular Pathogenesis of HPV
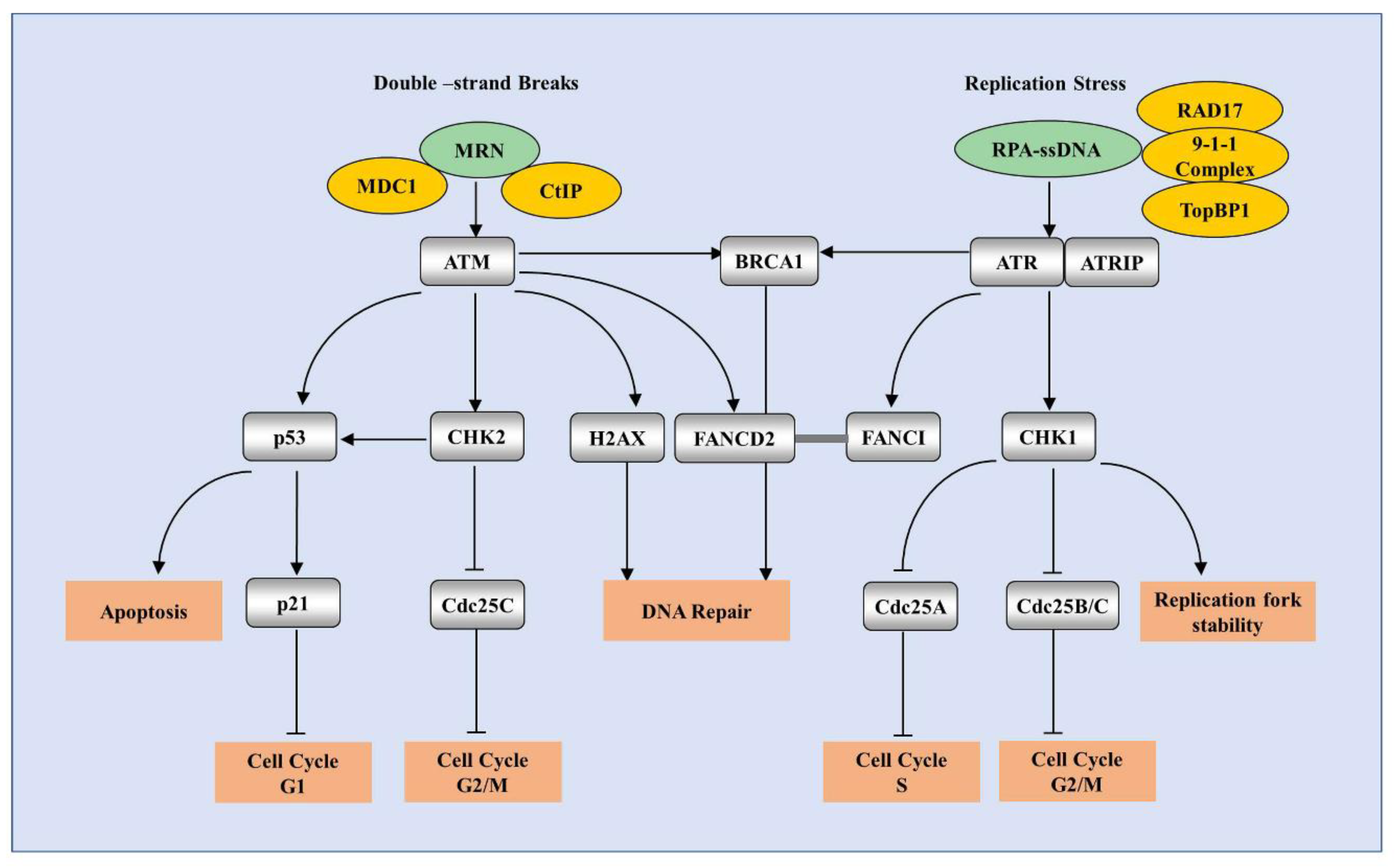
4.1. A Brief Overview of DDR
4.2. Activation of the ATM Pathway is Necessary for HPV Productive Replication
4.3. The Fanconi Anemia (FA) Pathway is Associated with HPV-Related Diseases
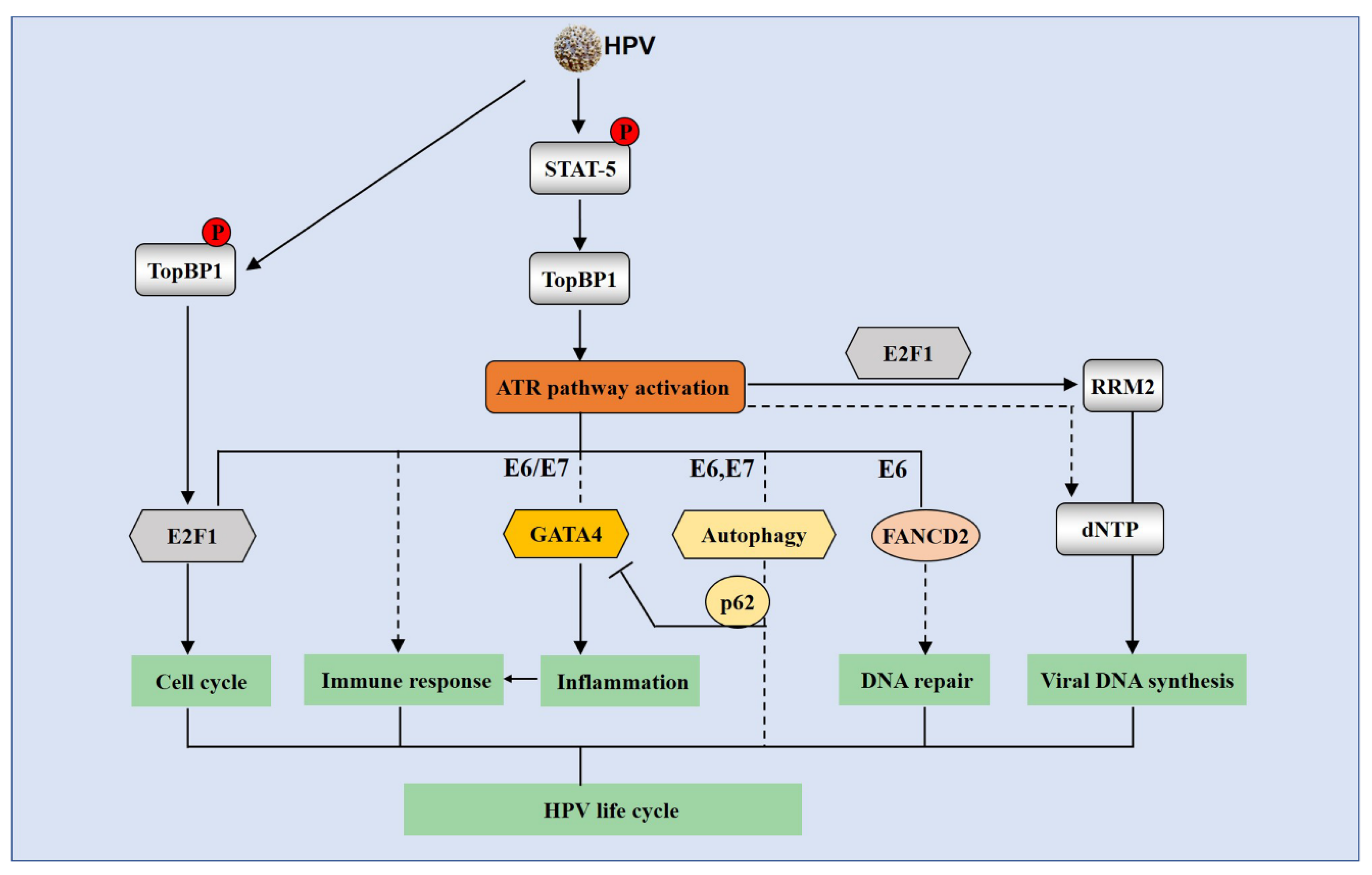
4.4. The ATR Pathway is Activated by HPV and Required for Efficient Viral Replication
5. Potential Therapeutic Prospects of ATR DDR Inhibitors Combined with Traditional Therapy for HPV-Associated Cancer
6. Summary and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- Chelmow, D. Practice bulletin No. 168: Cervical cancer screening and prevention. Obstet. Gynecol. 2016, 128, e111–e130. [Google Scholar]
- Patel, C.; Brotherton, J.M.; Pillsbury, A.; Jayasinghe, S.; Donovan, B.; Macartney, K.; Marshall, H. The impact of 10 years of human papillomavirus (HPV) vaccination in Australia: What additional disease burden will a nonavalent vaccine prevent? Eurosurveill 2018, 23. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Helmus, M.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J. Virol. 2013, 87, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Cheng, S.Q.; Iovane, A.; Laimins, L.A. STAT-5 regulates transcription of the topoisomerase iibeta-binding protein 1 (TopBP1) gene to activate the ATR pathway and promote human papillomavirus replication. MBIO 2015, 6, e02006–e02015. [Google Scholar] [CrossRef]
- Leonard, B.C.; Lee, E.D.; Bhola, N.E.; Li, H.; Sogaard, K.K.; Bakkenist, C.J.; Grandis, J.R.; Johnson, D.E. ATR inhibition sensitizes HPV(-) and HPV(+) head and neck squamous cell carcinoma to cisplatin. Oral. Oncol. 2019, 95, 35–42. [Google Scholar] [CrossRef]
- Zeng, L.; Beggs, R.R.; Cooper, T.S.; Weaver, A.N.; Yang, E.S. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol. Cancer 2017, 16, 591–600. [Google Scholar] [CrossRef]
- de Villiers, E.M. Cross-roads in the classification of papillomaviruses. Virology 2013, 445, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.F. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Slama, J.; Gonzalez, P.; Goodman, M.T.; Xia, N.; Kreimer, A.R.; Wu, T.; Hessol, N.A.; Shvetsov, Y.; Ortiz, A.P.; et al. Cervical determinants of anal HPV infection and high-grade anal lesions in women: A collaborative pooled analysis. Lancet Infect. Dis. 2019, 19, 880–891. [Google Scholar] [CrossRef]
- Mehanna, H.; Beech, T.; Nicholson, T.; El-Hariry, I.; McConkey, C.; Paleri, V.; Roberts, S. Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer-systematic review and meta-analysis of trends by time and region. Head Neck 2013, 35, 747–755. [Google Scholar] [CrossRef]
- Jeronimo, J.; Castle, P.E.; Temin, S.; Denny, L.; Gupta, V.; Kim, J.J.; Luciani, S.; Murokora, D.; Ngoma, T.; Qiao, Y.L. Secondary prevention of cervical cancer: ASCO resource-stratified clinical practice guideline. J. Glob. Oncol. 2016, 3, 635–657. [Google Scholar] [CrossRef]
- Huh, W.K.; Ault, K.A.; Chelmow, D.; Davey, D.D.; Goulart, R.A.; Garcia, F.A.; Kinney, W.K.; Massad, L.S.; Mayeaux, E.J.; Saslow, D.; et al. Use of primary high-risk human papillomavirus testing for cervical cancer screening: Interim clinical guidance. Gynecol. Oncol. 2015, 136, 178–182. [Google Scholar] [CrossRef]
- Burd, E.M. Human papillomavirus laboratory testing: The changing paradigm. Clin. Microbiol. Rev. 2016, 29, 291–319. [Google Scholar] [CrossRef]
- Saslow, D.; Solomon, D.; Lawson, H.W.; Killackey, M.; Kulasingam, S.L.; Cain, J.M.; Garcia, F.A.; Moriarty, A.T.; Waxman, A.G.; Wilbur, D.C.; et al. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. J. Low Genit. Tract. Dis. 2012, 16, 175–204. [Google Scholar] [CrossRef]
- Tovar, J.M.; Bazaldua, O.V.; Vargas, L.; Reile, E. Human papillomavirus, cervical cancer, and the vaccines. Postgrad. Med. 2008, 120, 79–84. [Google Scholar] [CrossRef] [PubMed]
- STD vaccine breakthrough. Cervarix would prevent human papilloma virus which can lead to cervical cancer; FDA approval anticipated. Health News 2005, 11, 6–7.
- Siddiqui, M.A.; Perry, C.M. Human papillomavirus quadrivalent (Types 6, 11, 16, 18) recombinant vaccine (gardasil). Drugs 2006, 66, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Moscicki, A.B.; Schiffman, M.; Burchell, A.; Albero, G.; Giuliano, A.R.; Goodman, M.T.; Kjaer, S.K.; Palefsky, J. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 2012, 5, F24–F33. [Google Scholar] [CrossRef]
- Roden, R.B.S.; Stern, P.L. Opportunities and challenges for human papillomavirus vaccination in cancer. Nat. Rev. Cancer 2018, 18, 240–254. [Google Scholar] [CrossRef]
- Shibata, T.; Lieblong, B.J.; Sasagawa, T.; Nakagawa, M. The promise of combining cancer vaccine and checkpoint blockade for treating HPV-related cancer. Cancer Treat. Rev. 2019, 78, 8–16. [Google Scholar] [CrossRef]
- Hebner, C.M.; Laimins, L.A. Human papillomaviruses: Basic mechanisms of pathogenesis and oncogenicity. Rev. Med. Virol. 2006, 16, 83–97. [Google Scholar] [CrossRef]
- Hughes, F.J.; Romanos, M.A. E1 protein of human papillomavirus is a DNA helicase/ATPase. Nucleic Acids Res. 1993, 21, 5817–5823. [Google Scholar] [CrossRef]
- Conger, K.L.; Liu, J.S.; Kuo, S.R.; Chow, L.T.; Wang, T.S. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human dna polymerase alpha/primase. J. Biol. Chem. 1999, 274, 2696–2705. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Steger, G.; Corbach, S. Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral E2 protein. J. Virol. 1997, 71, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Biryukov, J.; Myers, J.; McLaughlin-Drubin, M.; Griffin, H.; Milici, J.; Doorbar, J.; Meyers, C. Mutations in HPV18 E1^E4 impact virus capsid assembly, infectivity competence, and maturation. Viruses 2017, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Ely, S.; Sterling, J.; McLean, C.; Crawford, L. Specific interaction between HPV-16 E1–E4 and cytokeratins results in collapse of the epithelial cell intermediate filament network. Nature 1991, 352, 824–827. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef]
- Kivi, N.; Greco, D.; Auvinen, P.; Auvinen, E. Genes involved in cell adhesion, cell motility and mitogenic signaling are altered due to HPV 16 E5 protein expression. Oncogene 2008, 27, 2532–2541. [Google Scholar] [CrossRef]
- Liao, S.; Deng, D.; Zhang, W.; Hu, X.; Wang, W.; Wang, H.; Lu, Y.; Wang, S.; Meng, L.; Ma, D. Human papillomavirus 16/18 E5 promotes cervical cancer cell proliferation, migration and invasion in vitro and accelerates tumor growth in vivo. Oncol. Rep. 2013, 29, 95–102. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP Complex Functions as a Ubiquitin-Protein Ligase in the Ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [CrossRef]
- Felsani, A.; Mileo, A.M.; Paggi, M.G. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene 2006, 25, 5277–5285. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Meyers, C. Replication and assembly of human papillomaviruses. J. Dent. Res. 2009, 88, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J. Virol. 1998, 72, 142–150. [Google Scholar] [CrossRef]
- McKinney, C.C.; Hussmann, K.L.; McBride, A.A. The role of the DNA damage response throughout the papillomavirus life cycle. Viruses 2015, 7, 2450–2469. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Keratinocyte differentiation-dependent human papillomavirus gene regulation. Viruses 2017, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef]
- Morgan, E.L.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLoS Pathog. 2018, 14, e1006975. [Google Scholar] [CrossRef]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997, 11, 2090–2100. [Google Scholar] [CrossRef]
- He, W.; Staples, D.; Smith, C.; Fisher, C. Direct activation of cyclin-dependent kinase 2 by human papillomavirus E7. J. Virol. 2003, 77, 10566–10574. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.L.; Munger, K. Direct association of the HPV16 E7 oncoprotein with cyclin A/CDK2 and cyclin E/CDK2 complexes. Virology 2008, 380, 21–25. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Huh, K.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J. Virol. 2008, 82, 8695–8705. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.G.; Lee, D.; Kim, J.; Seo, T.; Choe, J. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. J. Biol. Chem. 2002, 277, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Xu, J.F.; Li, Y.; Andrade, J.; Hoover, P.; Kaminski, P.J.; Laimins, L.A. Topoisomerase IIβ-binding protein 1 activates expression of E2F1 and p73 in HPV-positive cells for genome amplification upon epithelial differentiation. Oncogene 2019, 38, 3274–3287. [Google Scholar] [CrossRef]
- Mighty, K.K.; Laimins, L.A. p63 is necessary for the activation of human papillomavirus late viral functions upon epithelial differentiation. J. Virol. 2011, 85, 8863–8869. [Google Scholar] [CrossRef]
- Hong, S.Y. DNA damage response is hijacked by human papillomaviruses to complete their life cycle. J. Zhejiang Univ. Sci. B 2017, 18, 215–232. [Google Scholar] [CrossRef]
- Wu, S.; Shi, Y.J.; Mulligan, P.; Gay, F.; Landry, J.; Liu, H.F.; Lu, J.; Qi, H.H.; Wang, W.J.; Nickoloff, J.A.; et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat. Struct. Mol. Biol. 2007, 14, 1165–1172. [Google Scholar] [CrossRef]
- Beishline, K.; Kelly, C.M.; Olofsson, B.A.; Koduri, S.; Emrich, J.; Greenberg, R.A.; Azizkhan-Clifford, J. Sp1 facilitates DNA double-strand break repair through a nontranscriptional mechanism. Mol. Cell. Biol. 2012, 32, 3790–3799. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Jin, S.; Fan, F.; Fan, W.; Tong, T.; Zhan, Q. Activation of the transcription factor Oct-1 in response to DNA damage. Cancer Res. 2000, 60, 6276–6280. [Google Scholar]
- Hong, S.Y.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef]
- Hong, S.Y.; Laimins, L.A. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef]
- Zhang, W.; Hong, S.Y.; Maniar, K.P.; Cheng, S.; Jie, C.; Rademaker, A.W.; Krensky, A.M.; Clayberger, C. KLF13 regulates the differentiation-dependent human papillomavirus life cycle in keratinocytes through STAT5 and IL-8. Oncogene 2016, 35, 5565–5575. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.K.; Li, Y.; Hafner, M.; Banerjee, N.S.; Tang, S.; Briskin, D.; Meyers, C.; Chow, L.T.; Xie, X.; et al. MicroRNAs are biomarkers of oncogenic human papillomavirus infections. Proc. Natl. Acad. Sci. USA 2014, 111, 4262–4267. [Google Scholar] [CrossRef] [PubMed]
- Melar-New, M.; Laimins, L.A. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 2010, 84, 5212–5221. [Google Scholar] [CrossRef]
- Gunasekharan, V.; Laimins, L.A. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J. Virol. 2013, 87, 6037–6043. [Google Scholar] [CrossRef]
- Nuovo, G.J.; Wu, X.; Volinia, S.; Yan, F.; di Leva, G.; Chin, N.; Nicol, A.F.; Jiang, J.; Otterson, G.; Schmittgen, T.D.; et al. Strong inverse correlation between microRNA-125b and human papillomavirus DNA in productive infection. Diagn. Mol. Pathol. 2010, 19, 135–143. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Yu, Y.; Riballo, E.; Douglas, P.; Walker, S.A.; Ye, R.; Harer, C.; Marchetti, C.; Morrice, N.; Jeggo, P.A.; et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 2006, 25, 3880–3889. [Google Scholar] [CrossRef]
- Brugmans, L.; Kanaar, R.; Essers, J. Analysis of DNA double-strand break repair pathways in mice. Mutat. Res. 2007, 614, 95–108. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-mediated end joining: A back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef]
- Bristol, M.L.; Das, D.; Morgan, I.M. Why human papillomaviruses activate the DNA damage response (DDR) and how cellular and viral replication persists in the presence of DDR signaling. Viruses 2017, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA replication and the host DNA damage response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Bhatti, S.; Kozlov, S.; Farooqi, A.A.; Naqi, A.; Lavin, M.; Khanna, K.K. ATM protein kinase: The linchpin of cellular defenses to stress. Cell. Mol. Life Sci. 2011, 68, 2977–3006. [Google Scholar] [CrossRef]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM Kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef]
- Concin, N.; Stimpfl, M.; Zeillinger, C.; Wolff, U.; Hefler, L.; Sedlak, J.; Leodolter, S.; Zeillinger, R. Role of p53 in G2/M cell cycle arrest and apoptosis in response to gamma-irradiation in ovarian carcinoma cell lines. Int. J. Oncol. 2003, 22, 51–57. [Google Scholar]
- Yazdi, P.T.; Wang, Y.; Zhao, S.; Patel, N.; Lee, E.Y.; Qin, J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002, 16, 571–582. [Google Scholar] [CrossRef]
- Taniguchi, T.; Garcia-Higuera, I.; Xu, B.; Andreassen, P.R.; Gregory, R.C.; Kim, S.T.; Lane, W.S.; Kastan, M.B. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 2002, 109, 459–472. [Google Scholar] [CrossRef]
- Garcia-Higuera, I.; Taniguchi, T.; Ganesan, S.; Meyn, M.S.; Timmers, C.; Hejna, J.; Grompe, M.; D‘Andrea, A.D. Interaction of the fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 2001, 7, 249–262. [Google Scholar] [CrossRef]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald 3rd, E.R.; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P. Identification of the FANCI Protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef]
- Ishiai, M.; Kitao, H.; Smogorzewska, A.; Tomida, J.; Kinomura, A.; Uchida, E.; Saberi, A.; Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T.; et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2008, 15, 1138–1146. [Google Scholar] [CrossRef]
- Lim, D.S.; Kim, S.T.; Xu, B.; Maser, R.S.; Lin, J.; Petrini, J.H.; Kastan, M.B. ATM phosphorylates p95 nbs1 in an S phase checkpoint pathway. Nature 2000, 404, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ting, N.S.; Zheng, L.; Chen, P.L.; Ziv, Y.; Shiloh, Y.; Lee, E.Y.; Lee, W.H. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 2000, 406, 210–215. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef]
- Chiang, C.M.; Ustav, M.; Stenlund, A.; Ho, T.F.; Broker, T.R.; Chow, L.T. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc. Natl. Acad. Sci. USA 1992, 89, 5799–5803. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef]
- Leverrier, S.; Bergamaschi, D.; Ghali, L.; Ola, A.; Warnes, G.; Akgul, B.; Blight, K.; Garcia-Escudero, R.; Penna, A.; Eddaoudi, A.; et al. Role of HPV E6 proteins in preventing UVB-induced release of pro-apoptotic factors from the mitochondria. Apoptosis 2007, 12, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Jong, J.E.; Jeong, K.W.; Shin, H.; Hwang, L.R.; Lee, D.; Seo, T. Human papillomavirus type 16 E6 protein inhibits DNA fragmentation via interaction with DNA fragmentation factor 40. Cancer Lett. 2012, 324, 109–117. [Google Scholar] [CrossRef]
- Shen, Y.; White, E. p53-dependent apoptosis pathways. Adv. Cancer Res. 2001, 82, 55–84. [Google Scholar] [CrossRef]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef]
- Hong, S.Y.; Dutta, A.; Laimins, L.A. The acetyltransferase Tip60 is a critical regulator of the differentiation-dependent amplification of human papillomaviruses. J. Virol. 2015, 89, 4668–4675. [Google Scholar] [CrossRef]
- Jha, S.; Vande Pol, S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 2010, 38, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Gunasekharan, V.; Satsuka, A.; Laimins, L.A. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with CTCF insulators. PLoS Pathog. 2015, 11, e1004763. [Google Scholar] [CrossRef]
- Satsuka, A.; Mehta, K.; Laimins, L.A. p38MAPK and MK2 pathways are important for the differentiation-dependent human papillomavirus life cycle. J. Virol. 2015, 89, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Laimins, L.A. FANCD2 binds human papillomavirus genomes and associates with a distinct set of DNA repair proteins to regulate viral replication. MBIO 2017, 8, e02340-16. [Google Scholar] [CrossRef]
- Taniguchi, T.; D‘Andrea, A.D. Molecular pathogenesis of Fanconi anemia: Recent progress. Blood 2006, 107, 4223–4233. [Google Scholar] [CrossRef] [PubMed]
- Spardy, N.; Duensing, A.; Charles, D.; Haines, N.; Nakahara, T.; Lambert, P.F.; Duensing, S. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J. Virol. 2007, 81, 13265–13270. [Google Scholar] [CrossRef]
- Hoskins, E.E.; Morreale, R.J.; Werner, S.P.; Higginbotham, J.M.; Laimins, L.A.; Lambert, P.F.; Brown, D.R.; Gillison, M.L.; Nuovo, G.J.; Witte, D.P.; et al. The fanconi anemia pathway limits human papillomavirus replication. J. Virol. 2012, 86, 8131–8138. [Google Scholar] [CrossRef]
- Hoskins, E.E.; Morris, T.A.; Higginbotham, J.M.; Spardy, N.; Cha, E.; Kelly, P.; Williams, D.A.; Wikenheiser-Brokamp, K.A.; Duensing, S.; Wells, S.I. Fanconi anemia deficiency stimulates HPV-associated hyperplastic growth in organotypic epithelial raft culture. Oncogene 2009, 28, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Kutler, D.I.; Wreesmann, V.B.; Goberdhan, A.; Ben-Porat, L.; Satagopan, J.; Ngai, I.; Huvos, A.G.; Giampietro, P.; Levran, O.; Pujara, K.; et al. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J. Natl. Cancer Inst. 2003, 95, 1718–1721. [Google Scholar] [CrossRef] [PubMed]
- Tomida, J.; Itaya, A.; Shigechi, T.; Unno, J.; Uchida, E.; Ikura, M.; Masuda, Y.J.; Matsuda, S. A novel interplay between the fanconi anemia core complex and atr-atrip kinase during DNA cross-link repair. Nucleic Acids Res. 2013, 41, 6930–6941. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Ohashi, E.; Takeishi, Y.; Ueda, S.; Tsurimoto, T. Interaction between Rad9-Hus1-Rad1 and TopBP1 activates ATR-ATRIP and promotes TopBP1 recruitment to sites of UV-damage. DNA Repair (Amst.) 2014, 21, 1–11. [Google Scholar] [CrossRef]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, R.S.; Brumbaugh, K.M.; Williams, J.M.; Sarkaria, J.N.; Cliby, W.A.; Shieh, S.Y.; Taya, Y.; Prives, C.; Abraham, R.T. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999, 13, 152–157. [Google Scholar] [CrossRef]
- Tibbetts, R.S.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000, 14, 2989–3002. [Google Scholar] [CrossRef]
- Singh, T.R.; Ali, A.M.; Paramasivam, M.; Pradhan, A.; Wahengbam, K.; Seidman, M.M.; Meetei, A.R. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res. 2013, 73, 4300–4310. [Google Scholar] [CrossRef]
- Collins, N.B.; Wilson, J.B.; Bush, T.; Thomashevski, A.; Roberts, K.J.; Jones, N.J.; Kupfer, G.M. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood 2009, 113, 2181–2190. [Google Scholar] [CrossRef]
- Andreassen, P.R.; D‘Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Jones, M.J.K.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J.; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef]
- Loo, Y.M.; Melendy, T. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J. Virol. 2004, 78, 1605–1615. [Google Scholar] [CrossRef]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef]
- Donaldson, M.M.; Mackintosh, L.J.; Bodily, J.M.; Dornan, E.S.; Laimins, L.A.; Morgan, I.M. An interaction between human papillomavirus 16 E2 and TopBP1 is required for optimum viral DNA replication and episomal genome establishment. J. Virol. 2012, 86, 12806–12815. [Google Scholar] [CrossRef] [PubMed]
- Boner, W.; Taylor, E.R.; Tsirimonaki, E.; Yamane, K.; Campo, M.S.; Morgan, I.M. A functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1. J. Biol. Chem. 2002, 277, 22297–22303. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Donaldson, M.M.; Dornan, E.S.; Wang, X.; Bristol, M.; Bodily, J.M.; Morgan, I.M. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2-mediated DNA replication. J. Virol. 2015, 89, 4980–4991. [Google Scholar] [CrossRef] [PubMed]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Simpson, D.A.; Zhou, Y.C.; Mitra, A.; Mitchell, D.L.; Cordeiro-Stone, M. Human papilloma virus type16 E6 deregulates CHK1 and sensitizes human fibroblasts to environmental carcinogens independently of its effect on p53. Cell Cycle 2009, 8, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Thatte, J.; Massimi, P.; Thomas, M.; Boon, S.S.; Banks, L.; Longnecker, R.M. The human papillomavirus E6 PDZ binding motif links DNA damage response signaling to E6 inhibition of p53 transcriptional activity. J. Virol. 2018, 92, e00465-18. [Google Scholar] [CrossRef]
- Chellappan, S.; Kraus, V.B.; Kroger, B.; Munger, K.; Howley, P.M.; Phelps, W.C.; Nevins, J.R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 4549–4553. [Google Scholar] [CrossRef]
- Anacker, D.C.; Aloor, H.L.; Shepard, C.N.; Lenzi, G.M.; Johnson, B.A.; Moody, C.A. HPV31 utilizes the ATR-Chk1 pathway to maintain elevated RRM2 levels and a replication-competent environment in differentiating Keratinocytes. Virology 2016, 499, 383–396. [Google Scholar] [CrossRef]
- Xu, J.F.; Liu, H.B.; Yang, Y.Q.; Wang, X.H.; Liu, P.C.; Zheng, Z.M. Genome-wide profiling of cervical RNA-binding proteins identifies human papillomavirus regulation of RNASEH2A expression by viral E7 and E2F1. MBIO 2019, 10, e02687–e02718. [Google Scholar] [CrossRef]
- Liu, K.; Graves, J.D.; Scott, J.D.; Li, R.; Lin, W.C. Akt switches TopBP1 function from checkpoint activation to transcriptional regulation through phosphoserine binding-mediated oligomerization. Mol. Cell. Biol. 2013, 33, 4685–4700. [Google Scholar] [CrossRef]
- Habiger, C.; Jager, G.; Walter, M.; Iftner, T.; Stubenrauch, F. Interferon kappa inhibits human papillomavirus 31 transcription by inducing Sp100 proteins. J. Virol. 2016, 90, 694–704. [Google Scholar] [CrossRef]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.K.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, W.G.; Zhao, Y. Autophagy substrate SQSTM1/p62 regulates chromatin ubiquitination during the DNA damage response. Autophagy 2017, 13, 212–213. [Google Scholar] [CrossRef]
- Liu, M.; Zeng, T.; Zhang, X.; Liu, C.; Wu, Z.; Yao, L.; Xie, C.; Xia, H.; Lin, Q.; Xie, L.; et al. ATR/Chk1 signaling induces autophagy through sumoylated RhoB-mediated lysosomal translocation of TSC2 after DNA damage. Nat. Commun. 2018, 9, 4139. [Google Scholar] [CrossRef]
- Hanning, J.E.; Saini, H.K.; Murray, M.J.; Caffarel, M.M.; van Dongen, S.; Ward, D.; Barker, E.M.; Scarpini, C.G.; Groves, I.J.; Stanley, M.A.; et al. Depletion of HPV16 early genes induces autophagy and senescence in a cervical carcinogenesis model, regardless of viral physical state. J. Pathol. 2013, 231, 354–366. [Google Scholar] [CrossRef]
- Zhang, B.; Song, Y.J.; Sun, S.Y.; Han, R.; Hua, C.T.; van der Veen, S.; Cheng, H. Human papillomavirus 11 early protein E6 activates autophagy by repressing AKT/mTOR and Erk/mTOR. J. Virol. 2019, 93, e00172-19. [Google Scholar] [CrossRef]
- Tingting, C.; Shizhou, Y.; Songfa, Z.; Junfen, X.; Weiguo, L.; Xiaodong, C.; Xing, X. Human papillomavirus 16E6/E7 activates autophagy via Atg9B and LAMP1 in cervical cancer cells. Cancer Med. 2019, 8, 4404–4416. [Google Scholar] [CrossRef]
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef]
- Akgul, B.; Kirschberg, M.; Storey, A.; Hufbauer, M. Human papillomavirus type 8 oncoproteins E6 and E7 cooperate in downregulation of the cellular checkpoint kinase-1. Int. J. Cancer 2019, 145, 797–806. [Google Scholar] [CrossRef]
- Edwards, T.G.; Bloom, D.C.; Fisher, C. The ATM and Rad3-Related (ATR) protein kinase pathway is activated by herpes simplex virus 1 and required for efficient viral replication. J. Virol. 2018, 92, e01884-17. [Google Scholar] [CrossRef]
- Luo, Y.; Lou, S.; Deng, X.; Liu, Z.; Li, Y.; Kleiboeker, S.; Qiu, J. Parvovirus B19 infection of human primary erythroid progenitor cells triggers ATR-Chk1 signaling, which promotes B19 virus replication. J. Virol. 2011, 85, 8046–8055. [Google Scholar] [CrossRef] [PubMed]
- Hammack, C.; Ogden, S.C.; Madden, J.C., Jr.; Medina, A.; Xu, C.C.; Phillips, E.; Son, Y.N.; Cone, A. Zika virus infection induces DNA damage response in human neural progenitors that enhances viral replication. J. Virol. 2019, 93, e00638-19. [Google Scholar] [CrossRef]
- Brestovitsky, A.; Nebenzahl-Sharon, K.; Kechker, P.; Sharf, R.; Kleinberger, T. The adenovirus E4orf4 protein provides a novel mechanism for inhibition of the DNA damage response. PLoS Pathog. 2016, 12, e1005420. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Patel, R.N.; Forrester, N.A.; Theil, K.; Groitl, P.; Stewart, G.S.; Taylor, A.M.; Morgan, I.M.; Dobner, T.; Grand, R.J.; et al. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc. Natl. Acad. Sci. USA 2010, 107, 12251–12256. [Google Scholar] [CrossRef] [PubMed]
- Koganti, S.; Hui-Yuen, J.; McAllister, S.; Gardner, B.; Grasser, F.; Palendira, U.; Tangye, S.G.; Freeman, A.F.; Bhaduri-McIntosh, S. STAT3 interrupts ATR-Chk1 signaling to allow oncovirus-mediated cell proliferation. Proc. Natl. Acad. Sci. USA 2014, 111, 4946–4951. [Google Scholar] [CrossRef]
- Hau, P.M.; Deng, W.; Jia, L.; Yang, J.; Tsurumi, T.; Chiang, A.K.; Huen, M.S.; Tsao, S.W. Role of ATM in the formation of the replication compartment during lytic replication of Epstein-Barr virus in nasopharyngeal epithelial cells. J. Virol. 2015, 89, 652–668. [Google Scholar] [CrossRef]
- Lung, R.W.; Hau, P.M.; Yu, K.H.; Yip, K.Y.; Tong, J.H.; Chak, W.P.; Chan, A.W.; Lam, K.H.; Lo, A.K.; Tin, E.K.; et al. EBV-encoded miRNAs target ATM-mediated response in nasopharyngeal carcinoma. J. Pathol. 2018, 244, 394–407. [Google Scholar] [CrossRef]
- Bose, S.; Yap, L.F.; Fung, M.; Starzcynski, J.; Saleh, A.; Morgan, S.; Dawson, C.; Chukwuma, M.B.; Maina, E.; Buettner, M.; et al. The ATM tumour suppressor gene is down-regulated in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2009, 217, 345–352. [Google Scholar] [CrossRef]
- Mehta, K.; Laimins, L.A. Human papillomaviruses preferentially recruit DNA repair factors to viral genomes for rapid repair and amplification. MBIO 2018, 9, e00064-18. [Google Scholar] [CrossRef]
- Banerjee, N.S.; Moore, D.; Parker, C.J.; Broker, T.R.; Chow, L.T. Targeting DNA damage response as a strategy to treat HPV infections. Int. J. Mol. Sci. 2019, 20, 5455. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Name | Cancer | Treatment | Clinical Trials | References |
|---|---|---|---|---|---|
| ATR Inhibitors | VE-822 (VX-970) | HPV(+) cells | / | - | Nakahara T, J Virol. 2015 |
| AZD6738 | HPV(+)HNSCC cells and patient-derived xenograft tumors | Radiation Cisplatin | - | Dillon MT, Clin Cancer Res. 2019 Leonard BC, Oral Oncol. 2019 | |
| BEZ235 | HPV(+)tonsillar and base of tongue squamous cell carcinoma, HPV anal carcinogenesis mouse model, HPV(+)HNSCC cells | FGFR inhibitor AZD4547, Radiation | - | Holzhauser S, Oncol Lett. 2019 Rademacher BL, Eur J Cancer Prev. 2019 Schötz U, Cancers (Basel). 2020 | |
| CHK1 Inhibitors | AZD7762 | HPV(+)HNSCC cells | / | - | Ghasemi F, Oncotarget. 2018 |
| LY2603618 | HPV(+)HNSCC cells | Wee1 inhibitor AZD1775 Radiation | - | Busch CJ, Radiother Oncol. 2017 | |
| MK-8776 | Cervical cancer cells HPV(+)HNSCC cells | Cisplatin PARP inhibitor niraparib Radiation | - | Banerjee NS, Int J Mol Sci. 2019 Molkentine JM, Int J Radiat Biol. 2020 | |
| LY2606368 | HPV(+)HNSCC cells | EGFR inhibitor cetuximab Radiation | Phase 1 NCT02555644 | Zeng L, Mol Cancer Ther. 2017 | |
| CCT244747 | HPV(+)HNSCC cells | Radiation paclitaxel | - | Barker HE, Mol Cancer Ther. 2016 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Y.; Hong, S. The Role of Ataxia Telangiectasia Mutant and Rad3-Related DNA Damage Response in Pathogenesis of Human Papillomavirus. Pathogens 2020, 9, 506. https://doi.org/10.3390/pathogens9060506
Luo Y, Hong S. The Role of Ataxia Telangiectasia Mutant and Rad3-Related DNA Damage Response in Pathogenesis of Human Papillomavirus. Pathogens. 2020; 9(6):506. https://doi.org/10.3390/pathogens9060506
Chicago/Turabian StyleLuo, Ying, and Shiyuan Hong. 2020. "The Role of Ataxia Telangiectasia Mutant and Rad3-Related DNA Damage Response in Pathogenesis of Human Papillomavirus" Pathogens 9, no. 6: 506. https://doi.org/10.3390/pathogens9060506
APA StyleLuo, Y., & Hong, S. (2020). The Role of Ataxia Telangiectasia Mutant and Rad3-Related DNA Damage Response in Pathogenesis of Human Papillomavirus. Pathogens, 9(6), 506. https://doi.org/10.3390/pathogens9060506