1. Introduction
One of the most significant threats to global food production and agriculture is the pathogenic oomycete
Phytophthora infestans (
P. infestans) [
1]. It was first described by Berkeley in 1846 and subsequently named
Phytophthora infestans by deBary in 1876, which aptly translates as “plant destroyer” [
2,
3]. Most infamously known for the Irish potato famine in 1845–1849, the pathogen caused incessant, devastating outbreaks of late potato blight resulting in the deaths of one million people and a further one million emigrating [
4]. The current global impact of
P. infestans is in excess of
$6.2 billion per annum due to crop loss and the use of fungicides [
5]. The necessity of fungicides to combat the spread of
P. infestans is evident and with resistant strains continually developing, designing novel fungicides to target this pathogen is of high importance.
The Irish Famine of the 1840s spurred the development of the first-ever pesticide to be adopted worldwide. Developed by French scientists, Pierre Millardet and Ulysse Gayon, the Bordeaux mixture was used to combat the
P. infestans blight. This fungicide is a mixture of copper sulphate and lime, which forms the active compound copper(II) hydroxide, stabilised by calcium sulphate. The emergence of this fungicidal treatment was imperative to the conservation of crops and averting hunger, therefore saving lives [
6]. The use of the Bordeaux mixture diminished through the emergence of less toxic and more targeted fungicides [
7].
The Bordeaux mixture was broadly used from the 1890s until it was superseded by the dithiocarbamate class of fungicides in the 1960s. The broad-spectrum fungicides were used due to their high biological and chemical activity, with the benefit of low production cost [
8]. Mancozeb (
1) and Propineb (
3) were two of the most widely used dithiocarbamate fungicides [
9,
10]. Both of these organosulfur fungicides have been found to break down into toxic metabolites as degradation begins during storage to form the respective toxic metabolites (
Figure 1) [
11,
12]. Ethylene thiourea
2 (R = H), one of the main metabolites of Mancozeb, has been shown to have many toxic effects in rats such as teratogenicity, developmental toxicity and promoting thyroid neoplasms [
11]. Propylene thiourea
4 (R = CH
3) from Proprineb has also been shown to affect the thyroid and the nervous system [
8]. Propineb (
2) has been banned by the European Commission and Mancozeb (
1) is now under scrutiny for its negative effects.
Following dithiocarbamates, phenylamide fungicides were developed in the 1970s. A significant advance in the control of
P. infestans is attributed to the phenylamides. Metalaxyl (
5) (
Figure 2) is one of the most popular phenylamide fungicides, however, seven years after the product was commercialised, the first resistant strains of
P. infestans were identified in Israel [
13]. By 1980, resistant isolates of
P. infestans were discovered outside of Israel, in Ireland, Switzerland and The Netherlands, demonstrating a need for further fungicidal development. To combat the growing resistance of
P. infestans for phenylamides, a mixture was formulated with other fungicides (e.g., Mancozeb,
1) which resulted in the reduction of resistant strains emerging [
14].
Approved by the EU in 2017, oxathiapiprolin (
6) has been used in more recent years as a targeted antioomycete fungicide (
Figure 2). It was designed and synthesised by Pasteris et al. in 2016 [
15] basing the structure around a piperidine-thiazole-carbonyl core. After numerous derivatives were synthesised and tested, the chemical structure of oxathiapiprolin (
6) exhibited the greatest control over
P. infestans with low toxicity in humans, birds and fish. The authors also investigated the mechanism of action of oxathiapiprolin and uncovered a novel target for oomycete inhibition. The compound binds to an oxysterol binding protein, although the role for this family is not very well understood. Oxathiapiprolin (
6) has been approved in many countries around the world and requires a smaller quantity to be used on crops [
16]. However, similarly to the phenylamides, oxathiapiprolin has already been found to be ineffective against emerging resistant strains of
Phytophthora capsici [
15,
17]. The current focus has turned towards the use of natural products as fungicides to replace the previous synthetic alternatives. Although the first report of antimicrobial natural products was in 1676, development has been slow for these compounds [
18]. Of prime interest for natural source fungicides is the essential oil, cinnamaldehyde.
Cinnamaldehyde (
7,
Figure 3) is a natural product extracted from the bark of the plant genus
Cinnamona. This conjugated aromatic compound has shown to inhibit the growth of bacteria, filamentous moulds and yeast [
19]. There are over 250 species of cinnamon plants and trees and are native to Sri Lanka and South India [
20]. Cinnamaldehyde was first isolated from cinnamon oil in 1834 by Dumas and Peligot [
21] and was first synthesised in 1854 by Chiozza [
22]. The essential oil is generally recognised as safe by the United States Food and Drug Administration and has been used in foods and medicines as flavourings for many years [
23]. It is also reported to possess many pharmacological activities such as antidiabetic and anti-inflammatory effects [
24,
25]. Extensive research has also been carried out on the antibacterial properties of cinnamaldehyde. The compound has demonstrated strong activity against many food-borne bacteria such as
Bacillus, Staphylococcus and
Enterobacter spp. and can therefore be used as a food preservative [
26,
27,
28].
Studies by Quintanilla and Soylu [
29,
30] were some of the first to demonstrate the effect essential oils have on
P. infestans and a follow up study by Ferhout et al. found that cinnamaldehyde (
7) expressed a greater inhibitory effect on fungal strains than thyme oil [
31]. Research by Raut determined cinnamaldehyde to be the most effective phenylpropanoid from plant origin against
Candida albicans biofilm formation, being effective at completely inhibiting mature biofilms and identifying its potential [
32]. Jantan et al. tested 14 essential oils against six dermatophytes, one filamentous fungus and five strains of yeasts, finding again that cinnamaldehyde possessed the strongest activity against all the different strains of fungi studied [
33].
From experimental evidence, it appears cinnamaldehyde (
7) inhibits cell wall biosynthesis, membrane function and some specific enzyme activities. However, specific cinnamaldehyde targets are still not well established. It is known that at lethal concentrations, the cell membrane is disrupted by cinnamaldehyde whereas at below lethal concentrations, it can inhibit ATPase and at low concentrations, it disrupts enzymes involved in cytokine action. It has been demonstrated in many studies that cinnamaldehyde (
7) interacts with microbial cell membranes and that it is capable of altering the membrane lipid profile [
34]. It is thought that the lipophilicity of cinnamaldehyde allows the molecule to partition through the lipid membrane of the cells and thus, disrupts bilayer structure. This renders the cell more permeable and prone to leakage of intracellular material causing cell death [
26]. At lethal concentrations, cinnamaldehyde acts as a noncompetitive inhibitor of cell wall synthesising enzymes leading to the prevention of cell wall formation [
35].
At sub-lethal concentrations, cinnamaldehyde inhibits transmembrane ATPase activity in
E. coli and
Listeria monocytogenes by entering into the cellular periplasm [
36]. This enzyme is essential for maintaining transmembrane electrochemical proton gradient, necessary for the maintenance of cellular pH and uptake of nutrients [
37]. At low concentration, cinnamaldehyde has been demonstrated to inhibit GDP dependent polymerisation, preventing cell division [
38]. In
P.parasitica var. nicotianae, Lu et al. found that cinnamaldehyde completely inhibited mycelial growth, sporangial formation, zoospore production and germination at concentrations ranging from about 10 μM to 50 μM [
39].
In a study of resistance of
Capsicum annuum (pepper plant) against root rot caused by
P.capsici, Li et al. discovered that resistant plants displayed increased transcription of genes involved in the phenylpropanoid biosynthesis pathway, especially those involved in cinnamaldehyde production [
40]. This suggests that cinnamaldehyde is a naturally occurring protective compound, whose production is enhanced in plant strains that are resistant to oomycete infections.
Transient Receptor Potential (TRP) ion channels play an important role in sensing damage to cells. Members of this channel superfamily, most closely related to the polycystic kidney disease (PKD) family, are encoded within the genomes of all oomycetes examined [
41]. Compounds that are susceptible to nucleophilic attack from cystine can cause TRP channel activation, disrupting Ca
2+ levels in the cell. Hu et al. demonstrated that the α, β-unsaturated bond in cinnamaldehyde is susceptible to nucleophilic attack from cystine, and therefore, modulates Ca
2+ homeostasis in
Phytophthora capsici (
P. capsici) [
42]. Intracellular free Ca
2+ is a universal second messenger in eukaryotic cells, regulates cellular processes such as sporulation, germination and growth and is important in every life-cycle stage [
41,
43]. The covalently bound cinnamaldehyde to cystine was postulated to activate the TRP channel leading to an efflux of Ca
2+ out of the cell and subsequently leads to cell death. To test their theory that the α, β-unsaturated bond in cinnamaldehyde is important, Hu monitored Ca
2+ levels using a cinnamaldehyde derivative without the α, β-unsaturated bond, hydrocinnamaldehyde (
8,
Figure 3). Treatment of
P. capsici with hydrocinnamaldehyde (
8) demonstrated no inhibitory effects of zoospore growth indicating that the Michael addition through the α, β-unsaturated bond of cinnamaldehyde was crucial for stimulating Ca
2+ efflux and growth inhibition of
P. capsici. Thus, although many studies exist on antimicrobial activity against several different groups of microorganisms, there is much to be learned about the mode of action of cinnamaldehyde 7 and the scope of its effects on P. infestans. As we set out to identify new agents against P. infestans, we initiated a broad study on the importance of the aldehyde aspect of cinnamaldehyde.
Previous cinnamaldehyde analogues that have been subjected to
P. infestans testing have primarily retained their aldehyde functionality and are substituted on the aromatic ring, examples of which are represented in
Figure 4 (
9–
13) [
44]. Separately, a recent study by Watamoto found a diverse set of compounds to demonstrate inhibitory effects on fungal strains, amongst which some of the structural features of our targets align [
45]. Two hit compounds from this screen (
14 and
15,
Figure 4) have shown strong fungicidal effects and to inhibit the metabolic activity of
C. albicans. Of these, Bay 11-7082 (
14) possesses an α, β-unsaturated bond similar to cinnamaldehyde and may act through a similar mechanism. Ellipticine (
15) has known antimicrobial and especially anticancer properties: testing at bacterial planktonic mode showed (
15) had averaged a minimal inhibitory concentration (MIC) of 8.45 µM across all six
Candida strains and hence is worthy of further investigation.
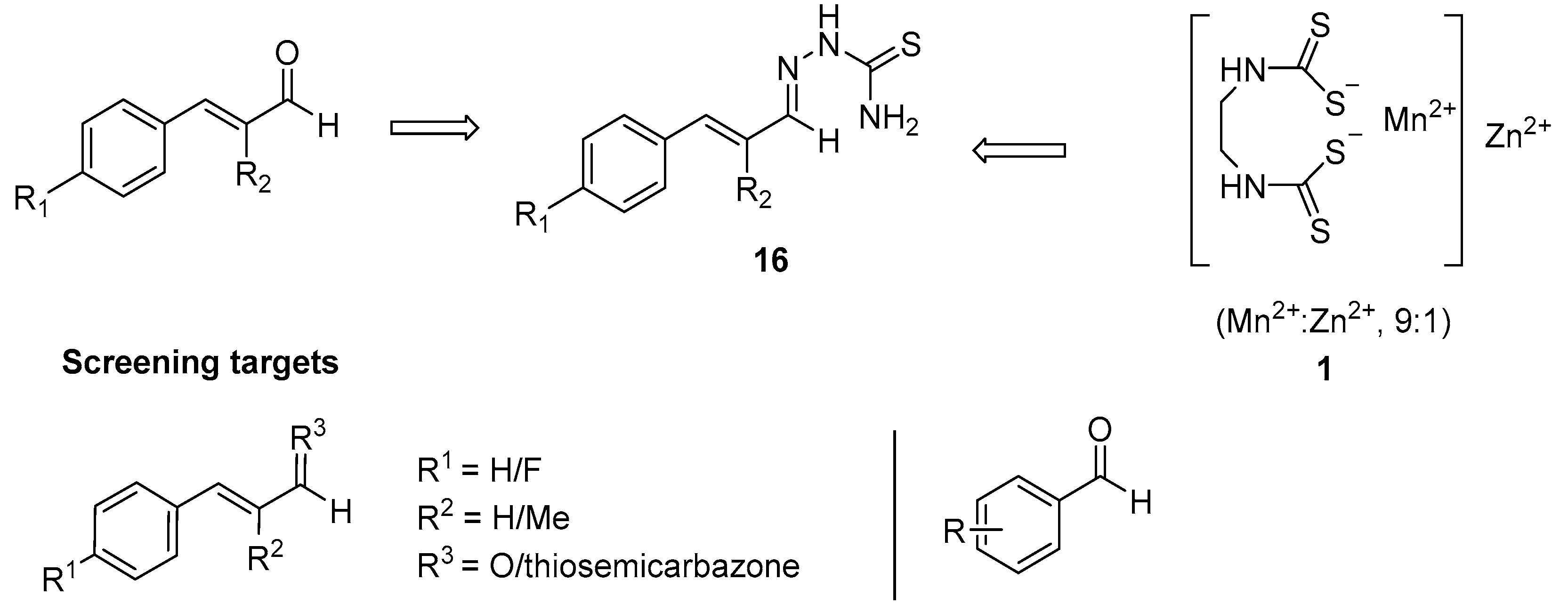
The aims of this study are to synthesise and evaluate cinnamaldehyde and aromatic aldehyde derivatives in assays of
P. infestans growth and zoospore production. Cinnamaldehyde derivatives will be accessed by specific synthetic modification at positions R
1, R
2 and R
3 represented in
Figure 5. 4-Fluorocinnamaldehyde (R
1 = F,
17) was chosen to probe the effect on electron density withdrawal on the Michael acceptor. At the R
2 position α-methylcinnamaldehyde (R
2 = CH
3,
18) will test steric effects as the methyl group can influence attack on this reactive element. Finally, the rationale is provided by the structure of Mancozeb (
1,
Figure 5) for the generation of a hybrid structure
16 and the related thiosemicarbazone at the R
3 position. Extension of the conjugated aldehyde will also be assessed to identify optimal chain length.
In order to probe the aldehyde functionality, a series of aromatic aldehydes and ellipticine aldehydes will also be assessed in the
P. infestans assay to screen for potential lead compounds for future development (
Figure 5). Given the experience of the group, of particular interest are the ellipticines which would offer a new framework for the inhibition of
P. infestans growth and the potential for overcoming resistance.
4. Materials and Methods
Solvents were distilled prior to use by the following methods: ethyl acetate was distilled from potassium carbonate; THF was freshly distilled from sodium and benzophenone and hexane was distilled prior to use. Organic phases were dried using anhydrous magnesium sulphate. Where room temperature is quoted in a method, this is within the temperature range 15–20 °C.
All commercial reagents were used without further purification unless otherwise stated. Alkyllithium reagents were titrated prior to use using the Gilman double titration procedure as follows; Two 100 mL conical flasks were prepared, the first one containing water (25 mL) and the second one containing dibromoethane (2 mL, stoppered with a SubaSeal under nitrogen with a provision for pressure release). The alkyllithium reagent (1 mL) was added via syringe to each flask [
56,
57]. The reaction with dibromoethane was more vigorous than that with water, and the flask was swirled during addition. Two drops of phenolphthalein were added to both flasks. The first flask was titrated against 0.1 M HCl until the purple colour disappeared. The SubaSeal was removed from the second flask and water (25 mL) was added, forming a biphasic mixture and was titrated as before. (Note: constant stirring was maintained to ensure mixing of both phases during titration). Titre value 1 represents the total base (alkyllithium and inorganic base) while titre 2 represents the free base (inorganic base not resulting from the alkyllithium). Thus, the molarity of the alkyllithium reagent was calculated using the following equation: molarity of alkyllithium (M) = [(titre 1−titre 2) × 0.1].
In reactions utilising alkyllithium and sodium hydride, all glassware was flame dried under nitrogen prior to use. All low-temperature reactions were carried out in a three-necked round-bottomed flask equipped with a low-temperature thermometer and the internal temperatures are quoted. Syringes were used to transfer small volumes of alkyllithium reagents while cannulation from the reagent bottle into a precalibrated addition funnel was used for larger volumes (>15 mL). Low-temperature reactions used the following cooling mixture: −100 °C, absolute ethanol and liquid nitrogen.
1H (300 MHz) and 13C (75.5 MHz) NMR spectra were recorded on a Bruker AVANCE 300 NMR spectrometer. 1H (600 MHz) and 13C (150.9 MHz) NMR spectra were recorded on a Bruker AVANCE III 600 NMR spectrometer equipped with a Bruker Dual C/H cryoprobe or a Bruker Broadband Observe H&F cryoprobe. All spectra were recorded at 300 K (26.9 °C) in deuterated dimethylsulfoxide (DMSO-d6) using DMSO-d6 as the reference peak or in deuterated chloroform (CDCl3) using trimethylsilane as an internal standard unless otherwise specified. Chemical shifts (δH and δC) are reported in parts per million (ppm) relative to the reference peak. Coupling constants (J) are expressed in Hertz (Hz). Splitting patterns in 1H spectra are designated as s (singlet), br s (broad singlet), d (doublet), t (triplet), br t (broad triplet), q (quartet), sept (septet), dd (doublet of doublets), ddd (doublet of doublet of doublets) and m (multiplet). Signal assignments were supported by COSY (correlation spectroscopy) or HMBC (Heteronuclear Multiple-Bond Correlation spectroscopy) experiments where necessary. 13C NMR spectra were assigned (aromatic C, CH, CH2, CH3) with the aid of DEPT (Distortionless Enhancement by Polarisation Transfer) experiments run in DEPT-90, DEPT-135 and DEPT-q modes. Specific assignments were made using HSQC (Heteronuclear Single Quantum Correlation) and HMBC (Heteronuclear Multiple-Bond Correlation) experiments. All spectroscopic data for known compounds were in agreement with those previously reported unless otherwise stated. Samples used for comparison (stacked plots) were run at equal concentrations (6–8 mg per 0.65 mL solvent). Carbon analysis is given for compounds that are novel or where full analysis has not been published in the literature.
Infrared spectra were recorded on a Bruker Tensor 37 FT-IR spectrophotometer interfaced with Opus version 7.2.139.1294 over a range of 400–4000 cm−1. An average of 16 scans was taken for each spectrum obtained with a resolution of 4 cm−1. Melting points were measured on a Uni-Melt Thomas Hoover capillary melting point apparatus and are uncorrected. Melting points or boiling points were not obtained for semi-solids or oils.
Thin Layer Chromatography (TLC) was carried out on precoated silica gel plates (Merck 60 F254). Visualisation was achieved by UV light detection (254 nm or 366 nm), Wet flash chromatography was carried out using Kieselgel silica gel 60, 0.040–0.063 mm (Merck).
Low-resolution mass spectra were recorded on a Waters Quattro Micro triple quadrupole spectrometer (QAA1202) in electron spray ionisation mode (ESI) using acetonitrile/water (1:1) containing 0.1% Formic acid as eluent. High-resolution mass spectrometry (HRMS) spectra were recorded on Waters Vion IMS (model no. SAA055K) in electron spray ionisation mode (ESI) using acetonitrile/water (1:1) containing 0.1% Formic acid as eluent.
Ethyl-(2E)-5-phenylpenta-2,4-dienoate 26
trans-Cinnamaldehyde 7 (0.100 g, 0.76 mmol) in dichloromethane (5 mL) was treated with (carbethoxymethylene)triphenylphosphorane 25 (0.267 g, 0.76 mmol) and allowed stir at room temperature for 1 h. The solvent was removed under reduced pressure and hexane was added. The triphenylphosphine oxide byproduct precipitated out as a pale pink solid and was isolated by vacuum filtration. The mother liquor was transferred into a round-bottomed flask and solvent was removed under reduced pressure. The crude product was purified by column chromatography eluting in hexane: ethyl acetate (100:0–99:1). The product eluted as a colourless oil (0.128 g, 83.5%). IR νmax/cm−1: 3062, 3019, 2982, 1723, 1178; δH (300 MHz, CDCl3): 1.31 (3H, t, J 7.1, CH2CH3), 4.22 (2H, q, J 7.1, CH2CH3), 5.98 (1H, d, J 15.2, H-2), 6.80-6.94 (2H, m, H-5, H-4), 7.24–7.50 (6H, m, H-3, ArH); m/z (ESI+) 202 [(M + H)+, 50%].
Ethyl-(2E,4E)-5-(4-fluorophenyl)penta-2,4-dienoate 27
4-Fuorocinnamaldehyde
17 (0.100 g, 0.33 mmol) in dichloromethane (5 mL) was treated with (carbethoxymethylene)triphenylphosphorane
25 (0.115 g, 0.33 mmol) and allowed stir at room temperature for 2 h. Analysis by TLC showed residual starting material so an additional portion of (carbethoxymethylene)triphenylphosphorane (0.010 mg, 0.03 mmol) was added and allowed stir overnight. The solvent was removed under reduced pressure and hexane was added. The white triphenylphosphine oxide byproduct precipitated out and was isolated by vacuum filtration. The mother liquor was transferred into a round-bottomed flask and solvent was removed under reduced pressure. The crude product was purified by column chromatography eluting in hexane: ethyl acetate (100:0–97:3). The product eluted as a colourless oil but solidified overnight to a white crystalline solid. (0.43 g, 64.5%). m.p. 41-43 °C (Lit. m.p. 44–46 °C) [
58]; IR ν
max/cm
−1: 2986, 1696, 1238, 1176; δ
H (300 MHz, CDCl
3): 1.31 (3H, t,
J 6.9, CH
2C
H3), 4.23 (2H, q,
J 6.9, C
H2CH
3), 5.98 (1H, d,
J 15.3, H-2), 6.77 (1H, dd,
J 15.3, 9.9, H-4), 6.86 (1H, d,
J 15.3, H-5), 7.04 (2H, t,
J 8.6, H-6′, H-2′), 7.36-7.47 (3H, m, H-5′, H-3′, H-3);
m/
z (ESI
+) 221 [(M + H)
+, 30%].
2-((E)-3-Phenylallylidene)hydrazine-1-carbothioamide 28
To a solution of
trans-cinnamaldehyde
7 (0.100 g, 0.76 mmol) in absolute ethanol (5 mL) was added thiosemicarbazide (0.069 mg, 0.76 mmol) and acetic acid (2 drops). The reaction mixture was heated to 80 °C and allowed stir for 45 min. The reaction mixture was cooled to 0 °C for 3 h without agitation. A precipitate formed and was isolated by vacuum filtration. The crude product was recrystallised from ethyl acetate: hexane to form pale yellow crystals (54 mg, 34.3%) m.p. 115–117 °C (Lit. m.p. 110–113 °C) [
59]; IR ν
max/cm
−1: 3413, 3255, 3155, 3028, 2981, 1610, 1590, 1536, 1372, 1291, 818; δ
H (300 MHz, CDCl
3): 6.47 (1H, br s, N
H2) 6.78 (1H, dd,
J 16.0, 9.0, H-2), 6.95 (1H, d,
J 16.0, H-3), 7.15 (1H, br s, N
H2), 7.30–7.40 (3H, m, H-2′, H-3′, H-4′), 7.45 (2H, dd,
J 8.1, 2.0, H-1′, H-5′), 7.75 (1H, d,
J 9.0, H-1), 10.08 (1H, s, CNN
H);
m/
z (ESI
+) 206 ((M + H)
+, 100%].
2-((E)-3-(4-Fluorophenyl)allylidene)hydrazine-1-carbothioamide 29
To a solution of 4-fluorocinnamaldehyde 17 (0.100 g, 0.67 mmol) in absolute ethanol (5 mL) was added thiosemicarbazide (0.060 g, 0.67 mmol) and acetic acid (2 drops). The reaction mixture was heated to 80 °C and allowed stir for 16 h. The reaction mixture was cooled to 0 °C for 3 h without agitation. A precipitate formed and was isolated by vacuum filtration. The crude product was recrystallised from ethyl acetate: hexane to form pure, pale yellow crystals (0.89 g, 59.7%) m.p. 144–146 °C; IR νmax/cm−1: 3386, 3249, 3149, 2985, 1610, 1600, 1528, 1505, 1475, 1286, 1156, 1082, 814; δH (300 MHz, CDCl3): 6.40 (1H, br s, NH2), 6.72 (1H, dd, J 16.0, 9.0, H-3), 6.90 (1H, d, J 16.0, H-2), 7.05 (2H, t, J 8.6, H-2′, H-6′), 7.13 (1H, br s, NH2), 7.43 (2H, dd, J 8.6, 5.4, H-5′, H-3′), 7.72 (1H, d, J 9.0, H-1), 9.95 (1H, br s, CNNH); δC (75.5 MHz, DMSO-d6): 79.63 (CH), 116.27 (CH, JC-F 21.7, aromatic CH), 125.41 (CH, aromatic CH), 125.44 (CH, aromatic CH), 129.47 (d, CH JC-F 8.6, aromatic CH), 133.02 (d, C, JC-F 3.3, aromatic C), 138.09 (CH), 145.11 (CH), 162.75 (d, C, JC-F 247.1, CF), 178.19 (C, CS); m/z (ESI+) 224 [(M + H)+, 100%]; HRMS (ESI+): Exact mass calculated for [C10H11FN3S]+ 224.0652. Found 224.0651.
2-Methyl-3-phenylallylidene hydrazine-1-carbothioamide 30
To a solution of α-methylcinnamaldehyde
18 (0.100 g, 0.68 mmol) in absolute ethanol (5 mL) was added thiosemicarbazide (0.062 g, 0.68 mmol) and acetic acid (2 drops). The reaction mixture was heated to 80 °C and allowed stir for 48 h. The reaction mixture was cooled to 0 °C for 3 h without agitation. A precipitate formed and was isolated by vacuum filtration. The crude product was recrystallised from ethyl acetate: hexane to form a paper-like white solid (0.76 g, 51.0%) m.p. 173–175 °C (Lit. m.p. 170–172 °C) [
60]; IR ν
max/cm
−1: 3423, 3241, 3149, 2965, 1602, 1508, 819; δ
H (300 MHz, CDCl
3): 2.12 (3H, s, CH
3-2), 6.39 (1H, br s, N
H2), 6.80 (1H, s, H-3), 7.14 (1H, br s, N
H2), 7.27-7.41 (5H, m, Ar
H), 7.69 (1H, s, H-1), 9.73 (1H, br s, CNN
H);
m/
z (ESI
+) 220 [(M + H)
+, 100%].
5,6,11-Trimethyl-6H-pyrido [4,3-b]carbazole 31
5,11-Dimethyl-6
H-pyrido[4,3
b]carbazole
15 (1.50 g, 6.10 mmol) in anhydrous DMF (20 mL) was treated with NaH (0.375 g, 60% dispersion in mineral oil 9.4 mmol,) at 0 °C under nitrogen and allowed stir for 30 min, forming a deep red suspension. Iodomethane (0.39 mL, 6.20 mmol) was added and the reaction was allowed warm to room temperature and stir overnight. The reaction mixture was cooled to 0 °C and water (100 mL) was added producing a pale orange precipitate. The yellow product was isolated by vacuum filtration and washed with water (150 mL) and did not require further purification (1.46 g, 91.8%). m.p 205–206 °C (Lit. m.p 202–204 °C) [
61]; IR ν
max/cm
−1: 2921, 2852, 1588, 1476, 1449, 1241, 1102, 806; δ
H (300 MHz, DMSO-
d6): 3.03 (3H, s, CH
3-5), 3.19 (3H, s, CH
3-11), 4.13 (3H, s, NCH
3), 7.31 (1H, overlapping ddd,
J 8.0, 6.4, 1.5, H-9), 7.58 (1H, dd,
J 8.0, 1.5, H-8), 7.62 (1H, d,
J 1.5, H-7), 7.99 (1H, d,
J 6.0, H-4), 8.37 (1H, d,
J 8.0, H-10), 8.45 (1H, d,
J 6.0, H-3), 9.68 (1H, s, H-1);
m/
z (ESI
+) 261 [(M + H)
+, 100%].
6-Ethyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole 32
5,11-Dimethyl-6H-pyrido[4,3-b]carbazole 15 (3.33 g, 13.5 mmol) in anhydrous DMF (60 mL), at 0 °C under nitrogen, was treated with sodium hydride (0.812 g, 20.3 mmol, 60% dispersion in mineral oil) and left to stir for 30 min forming a red suspension. Iodoethane (1.14 mL, 2.22 g, 14.2 mmol) was added and the reaction was allowed to warm to room temperature and was left to stir for a total of 72 h. The reaction mixture was cooled on ice and water (200 mL) was added. The aqueous layer was extracted with dichloromethane-methanol 90:10 (5 × 50 mL). The combined organic extracts were washed with water (5 × 50 mL) and brine (1 × 50 mL), dried over magnesium sulphate, filtered and the solvent removed under reduced pressure. Purification by column chromatography eluting with dichloromethane-methanol (99:1) gave the product as a yellow solid (2.35 g, 63.4%). m.p. 164–165 °C; νmax/cm−1 (KBr): 3020, 2966, 2926, 1596, 1588, 1475, 1395, 1382, 1346, 1232, 741; δH (600 MHz, DMSO-d6): 1.33 [3H, t, J 7.1, N(6)CH2CH3], 2.84 [3H, s, C(5)CH3], 3.03 [3H, s, C(11)CH3], 4.50 [2H, q, J 7.1, N(6)CH2CH3], 7.26 [1H, t, J 7.4, C(9)H], 7.53–7.58 [2H, m, C(7)H, C(8)H], 7.92 [1H, d, J 6.1, C(4)H], 8.26 [1H, d, J 7.8, C(10)H], 8.41 [1H, d, J 6.0, C(3)H], 9.58 [1H, s, C(1)H]; δC (150.9 MHz, DMSO-d6): 13.4 [CH3, C(5)CH3], 14.7 [CH3, C(11)CH3], 15.4 [CH3, N(6)CH2CH3], 40.2 [CH2, N(6)CH2CH3], 108.7 (C, aromatic C), 109.4 (CH, aromatic CH), 116.3 (CH, aromatic CH), 120.1 (CH, aromatic CH), 122.4 (C, aromatic C), 123.3 (C, aromatic C), 124.2 (CH, aromatic CH), 124.5 (C, aromatic C), 127.7 (CH, aromatic CH), 128.8 (C, aromatic C), 134.0 (C, aromatic C), 140.3 (C, aromatic C), 141.2 (CH, aromatic CH), 144.1 (C, aromatic C), 149.9 (CH, aromatic CH); m/z (ESI+): 275 [(M + H)+ 100%]; HRMS (ESI+): Exact mass calculated for C19H19N2+ 275.1548. Found 275.1547.
6-Isopropyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole 33
5,11-Dimethyl-6H-pyrido[4,3-b]carbazole 15 (2.31 g, 9.37 mmol) was added portion-wise to a suspension of sodium hydride (0.75 g, 60% dispersion in mineral oil, equivalent to 0.45 g sodium hydride, 18.8 mmol) in dimethylformamide (10 mL) forming a deep red suspension which was stirred at room temperature for 45 min under nitrogen. 2-Iodopropane (1.10 mL, 1.90 g, 11.2 mmol) in dimethylformamide (5 mL) was added dropwise and the reaction was stirred for 48 h. The reaction mixture was cooled on ice and water (50 mL) added. The aqueous layer was extracted with dichloromethane—Methanol 90:10 (3 × 50 mL). A precipitate in the aqueous layer which would not extract was filtered and found to be unreacted ellipticine (402 mg). Combined organic layers were washed with water (5 × 20 mL) and brine (1 × 30 mL), dried over magnesium sulphate and solvent removed under reduced pressure. Purification by column chromatography eluting with dichloromethane – methanol 99:1 gave the product as a yellow solid (0.96 g, 35.5%). m.p. 168–170 °C; νmax/cm−1 (KBr): 3435, 3079, 2970, 2933, 1598, 1587, 1469, 1390, 1367, 1345, 1288, 1236, 1104, 804, 741; δH (600 MHz, DMSO-d6): 1.59 [6H, d, J 6.9, N(6)CH(CH3)2], 2.87 [3H, s, C(5)CH3], 3.15 [3H, s, C(11)CH3], 5.32 [1H, sept, J 7.0, N(6)CH(CH3)2], 7.28 [1H, overlapping ddd, J 7.8, 7.2, 0.5, C(9)H], 7.51 [1H, overlapping ddd, J 8.2, 7.2, 1.0, C(8)H], 7.77 [1H, d, J 8.2, C(7)H], 7.92 [1H, d, J 6.0, C(4)H], 8.34 [1H, d, J 7.8, C(10)H], 8.43 [1H, d, J 5.8, C(3)H], 9.66 [1H, s, C(1)H]; δH (600 MHz, DMSO-d6): 14.8 [CH3, C(11)CH3], 15.3 [CH3, C(5)CH3], 21.4 [2 × CH3, N(6)CH(CH3)2], 49.6 [CH, N(6)CH(CH3)2], 109.6 (C, aromatic C), 113.6 (CH, aromatic CH), 116.6 (CH, aromatic CH), 120.2 (CH, aromatic CH), 122.9 (C, aromatic C), 124.5 (CH, aromatic CH), 124.9 (C, aromatic C), 125.3 (C, aromatic C), 127.0 (CH, aromatic CH), 128.3 (C, aromatic C), 134.6 (C, aromatic C), 141.4 (CH, aromatic CH), 143.1 (C, aromatic C), 143.2 (C, aromatic C), 149.9 (CH, aromatic CH); m/z (ESI+): 289 [(M + H)+, 100%]; HRMS (ESI+): Exact mass calculated for C20H21N2+ 289.1705. Found 289.1707.
5,6,11-Trimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 34
5,6,11-Trimethyl-6
H-pyrido[4,3-
b]carbazole
31 (0.600 g, 2.33 mmol) was dissolved in trifluoroacetic acid (30 mL) and hexamethylenetetramine (3.24 g, 23.41 mmol) added portion-wise. The reaction was heated to reflux for 1 h. Upon cooling to room temperature, the mixture was cooled to 0 °C and poured into water (200 mL). Solid sodium bicarbonate was added until neutralised and the mixture was extracted with dichloromethane-methanol (150 mL × 4, 9:1). The combined organic layers were washed with water (2 × 100 mL) and brine (2 × 100 mL), dried, filtered and solvent removed under reduced pressure to yield an orange solid (0.53 g, 81.0%). m.p. 219–222 °C (Lit. m.p. 222-223 °C) [
55,
62]; IR ν
max/cm
−1: 3214, 2922, 2852, 1674, 1586, 1461, 1380, 1356, 1099, 983; δ
H (300 MHz, DMSO-
d6): 3.01 (3H, s, CH
3-5), 3.21 (3H, s, CH
3-11), 4.16 (3H, s, NCH
3), 7.75 (1H, d,
J 8.7, H-7), 8.01 (1H, d,
J 5.8, H-4), 8.07 (1H, dd,
J 8.7, 1.5, H-8), 8.49 (1H, d,
J 5.8, H-3), 8.78 (1H, d,
J 1.5, H-10), 9.70 (1H, s, H-1), 10.10 (1H, s, C
HO);
m/
z (ESI
+) 289 [(M + H)
+, 100%].
6-Ethyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 35
6-Ethyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole 32 (1.00 g, 3.65 mmol) was stirred in trifluoroacetic acid (40 mL) and hexamethylenetetramine (5.11 g, 36.5 mmol) was added portion-wise. The reaction was heated to reflux for 40 min. Upon cooling the reaction mixture was cooled on ice and poured into water (250 mL). The reaction mixture was neutralised using solid sodium bicarbonate [Caution: vigorous reaction] and extracted with dichloromethane-methanol 90:10 (3 × 100 mL). The combined organic extracts were washed with water (2 × 100 mL) and brine (1 × 100 mL), dried over magnesium sulphate, filtered and the solvent removed under reduced pressure to give the product as an orange solid which was used without further purification (1.00 g, 91.0%). m.p. 192–196 °C; νmax/cm−1 (KBr): 2970, 2929, 2720, 1682, 1589, 1452, 1381, 1315, 1235, 1100, 809; δH (500 MHz, DMSO-d6): 1.42 [3H, t, J 6.9, N(6)CH2CH3], 2.95 [3H, s, C(5)CH3], 3.19 [3H, s, C(11)CH3], 4.66 [2H, q, J 6.8, N(6)CH2CH3], 7.78 [1H, d, J 8.5, C(7)H], 8.03 [1H, d, J 5.9, C(4)H], 8.08 [1H, d, J 8.4, C(8)H], 8.49 [1H, d, J 5.6, C(3)H], 8.77 [1H, s, C(10)H], 9.79 [1H, s, C(1)H], 10.10 [1H, s, C(9)CHO]; δC (125.8 MHz, DMSO-d6): 13.4 [CH3, C(5)CH3], 14.9 [CH3, C(11)CH3], 15.5 [CH3, N(6)CH2CH3], 40.7 [CH2, N(6)CH2CH3], 109.9 (CH, aromatic CH), 110.3 (C, aromatic C), 116.6 (CH, aromatic CH), 122.9 (C, aromatic C), 123.3 (C, aromatic C), 123.9 (C, aromatic C), 127.1 (CH, aromatic CH), 128.9 (CH, aromatic CH), 129.3 (C, aromatic C), 129.7 (C, aromatic C), 134.5 (C, aromatic C), 140.6 (C, aromatic C), 141.9 (CH, aromatic CH), 147.9 (C, aromatic C), 150.2 (CH, aromatic CH), 192.4 [C, C(9)CHO]; m/z (ESI+): 303 [(M + H)+, 100%]; HRMS (ESI+): Exact mass calculated for C20H19N2O+ 303.1497. Found 303.1485.
6-Isopropyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 36
6-Isopropyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole 33 (0.81 g, 2.82 mmol) was stirred in trifluoroacetic acid (40 mL) and hexamethylenetetramine (3.96 g, 28.2 mmol) was added portion-wise. The reaction was heated to reflux for 40 min. Upon cooling the reaction mixture was cooled on ice and poured into water (250 mL). The reaction mixture was neutralised using solid sodium bicarbonate [Caution: vigorous reaction] and extracted with dichloromethane-methanol 90:10 (3 × 100 mL). The combined organic extracts were washed with water (2 × 100 mL) and brine (1 × 100 mL), dried over magnesium sulphate, filtered and the solvent removed under reduced pressure to give the product as an orange solid which was used without further purification (0.836 g, 93.7%). m.p. 200–205 °C; νmax/cm−1 (KBr): 3412, 2969, 2929, 1683, 1588, 1574, 1386, 1321, 1286, 1240, 1192, 1140, 1104, 812; δH (600 MHz, DMSO-d6): 1.67 [6H, d, J 7.0, N(6)CH(CH3)2], 2.94 [3H, s, C(5)CH3], 3.24 [3H, s, C(11)CH3], 5.45 [1H, sept, J 6.9, N(6)CH(CH3)2], 7.97 [1H, d, J 8.6, C(7)H], 8.01 [1H, d, J 6.1, C(4)H], 8.04 [1H, dd, J 8.6, 1.4, C(8)H], 8.50 [1H, d, J 6.0, C(3)H], 8.84 [1H, d, J 1.1, C(10)H], 9.73 [1H, s, C(1)H], 10.11 [1H, s, C(9)CHO]; δC (150.9 MHz, DMSO-d6): 14.9 [CH3, C(11)CH3], 15.3 [CH3, C(5)CH3], 21.3 [2 × CH3, N(6)CH(CH3)2], 50.0 [CH, N(6)CH(CH3)2], 110.8 (C, aromatic C), 113.6 (CH, aromatic CH), 116.9 (CH, aromatic CH), 123.2 (C, aromatic C), 124.1 (C, aromatic C), 125.2 (C, aromatic C), 127.2 (CH, aromatic CH), 128.0 (CH, aromatic CH), 129.1 (C, aromatic C), 129.3 (C, aromatic C), 135.0 (C, aromatic C), 141.9 (CH, aromatic CH), 143.0 (C, aromatic C), 147.1 (C, aromatic C), 150.1 (CH, aromatic CH), 192.6 [C, C(9)CHO]; m/z (ESI+): 317 [(M + H)+, 100%]; HRMS (ESI+): Exact mass calculated for C21H21N2O+ 317.1654. Found 317.1651.
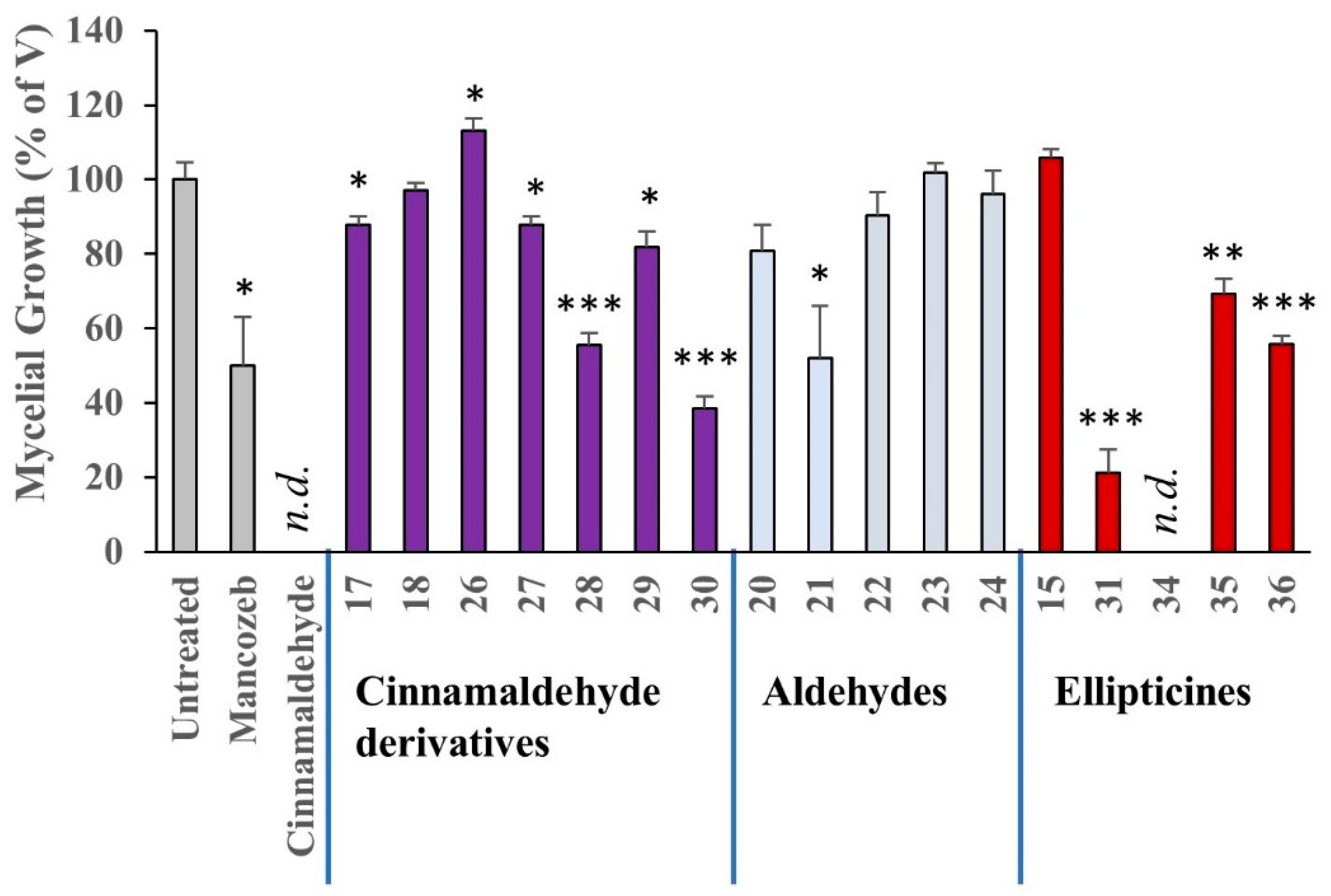
Phytophthora infestans Mycelial Growth Assays
Phytophthora infestans strain 88,069 (mating type A1) was originally obtained from Professor Sophien Kamoun (The Sainsbury Laboratory, Norwich, UK) and maintained on RyeA agar at 20 °C in the dark [
63]. For mycelial growth assay, compounds were dissolved in DMSO in a vial and 15 mL Rye B agar added, at a temperature of approximately 50 °C (the final concentration of DMSO was 0.1%). The vial was shaken to distribute the contents and the mixture was then poured into a clean, sterile 10 cm Petri dish. A 10 mm plug of
P. infestans mycelium was placed in the centre of the dish, which was then sealed with parafilm. The experiments were carried out in triplicate at 25 µM (unless otherwise stated), stored at 20 °C in the dark and checked periodically for mycelial growth.
Phytophthora infestans Zoosporogenesis and Sporangiogenesis Assays
These were performed essentially as described by Lu et al. [
39]. Following two weeks of culture on Rye B agar, sporangia were collected from plates in 5 mL of modified Petri’s solution (MPS, 5 mM CaCl
2, 1 mM MgSO
4, 1 mM KH
2PO
4 and 0.8 mM KCl), by scraping with a scalpel blade and were transferred to a 50 mL sterile tube. Plates were washed with another 5 mL of MPS and this was combined with the first 5 mL. To initiate zoospore release, sporangia were place on ice, in the dark, for 3h. Sporangia or zoospores were counted by brightfield microscopy using a 10× objective and a haemocytometer, in duplicate.
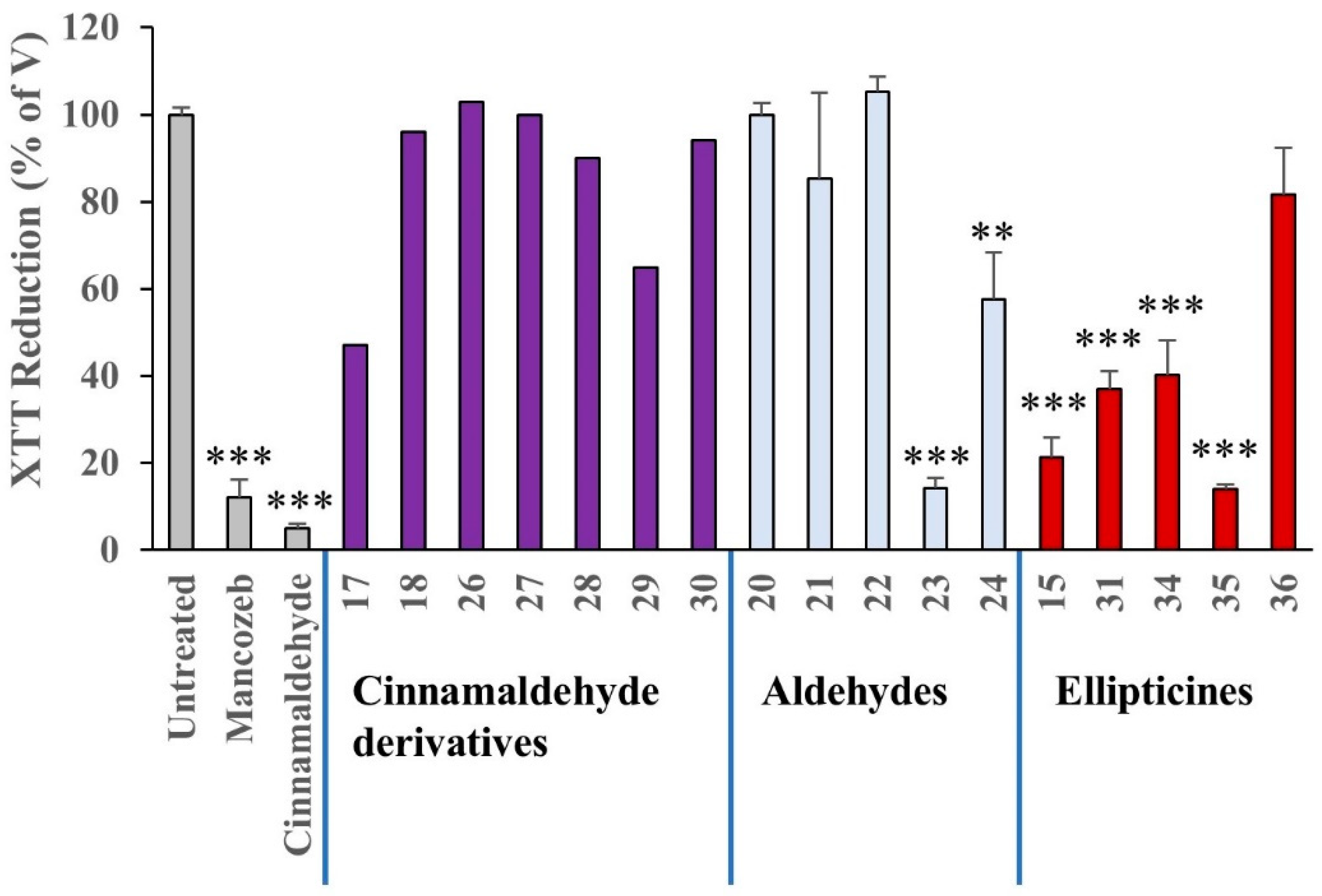
XTT reduction assay
Human embryonic kidney-293T (HEK-293T) cells were obtained from LGC Standards (Middlesex, UK) and were maintained in Dulbecco’s Modified Eagle’s Medium containing 10% foetal bovine serum, 100 units/mL penicillin and 100 μg/mL streptomycin, at 37 °C in a humidified atmosphere of 5% CO
2/95% air. For assays, cells were subcultured on 96-well microtitre plates at a density of 5 × 10
4 cells/well, in a volume of 50 mL. Following overnight culture, 50 mL of each test compound at twice their final concentration (50 mM for all apart from cinnamaldehyde (2 mM) and Mancozeb (200 μM)) was added and incubated for another 24 h. Cells were incubated with 100 mL XTT reagents (Sigma-Aldrich, Ireland) for an additional 4 h, after which, the absorbance of the extracellular formazan reduction product was measured at a wavelength of of 490 nm, with a reference wavelength of 675 nm [
53,
54], using a microtitre plate spectrophotometer.
Statistical Methods
Numerical data were statistically compared using one-way analysis of variance (ANOVA) and Tukey’s post hoc test (using ezANOVA software, available at:
https://people.cas.sc.edu/rorden/ezanova/index.html.) A probability threshold of
p < 0.05 was taken as statistically significant. Generation of histograms and linear regression analyses were performed using Microsoft Excel.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















