4. Materials and Methods
4.1. General Procedures
Solvents were distilled prior to use by the following methods: Ethyl acetate was distilled from potassium carbonate; THF was freshly distilled from sodium and benzophenone and hexane was distilled prior to use. Organic phases were dried using anhydrous magnesium sulfate.
All commercial reagents were used without further purification unless otherwise stated. Alkyllithium reagents were titrated prior to use using the Gilman double titration procedure as follows: Two 100 mL conical flasks were prepared, the first one containing water (25 mL) and the second one containing dibromoethane (2 mL, stoppered with a SubaSeal under nitrogen with a provision for pressure release). The alkyllithium reagent (1 mL) was added via syringe to each flask [
43,
44]. The reaction with dibromoethane was more vigorous than that with water, and the flask was swirled during addition. Two drops of phenolphthalein were added to both flasks. The first flask was titrated against 0.1 M HCl until the purple colour disappeared. The SubaSeal was removed from the second flask and water (25 mL) was added, forming a biphasic mixture and was titrated as before. (Note: Constant stirring was maintained to ensure mixing of both phases during titration). Titre value 1 represents the total base (alkyllithium and inorganic base) while titre 2 represents the free base (inorganic base not resulting from the alkyllithium). Thus, the molarity of the alkyllithium reagent was calculated using the following equation: molarity of alkyllithium (M) = [(titre 1 – titre 2) × 0.1].
In reactions utilising alkyllithium and sodium hydride, all glassware was flame dried under nitrogen prior to use. All low temperature reactions were carried out in a three-necked round bottomed flask equipped with a low temperature thermometer and the internal temperatures are quoted. Syringes were used to transfer small volumes of alkyllithium reagents while cannulation from the reagent bottle into a pre-calibrated addition funnel was used for larger volumes (>15 mL). Low temperature reactions used the following cooling mixture: −100 °C, absolute ethanol and liquid nitrogen.
1H (300 MHz) and 13C (75.5 MHz) NMR spectra were recorded on a Bruker AVANCE 300 NMR spectrometer. 1H (600 MHz) and 13C (150.9 MHz) NMR spectra were recorded on a Bruker AVANCE III 600 NMR spectrometer equipped with a Bruker Dual C/H cryoprobe or a Bruker Broadband Observe H&F cryoprobe. All spectra were recorded at 300 K (26.9 °C) in deuterated dimethylsulfoxide (DMSO-d6) using DMSO-d6 as the reference peak or in deuterated chloroform (CDCl3) using trimethylsilane as an internal standard unless otherwise specified. Chemical shifts (δH and δC) are reported in parts per million (ppm) relative to the reference peak. Coupling constants (J) are expressed in Hertz (Hz). Splitting patterns in 1H spectra are designated as s (singlet), br s (broad singlet), d (doublet), t (triplet), br t (broad triplet), q (quartet), sept (septet), dd (doublet of doublets), ddd (doublet of doublet of doublets) and m (multiplet). Signal assignments were supported by COSY (correlation spectroscopy) or HMBC (Heteronuclear Multiple-Bond Correlation spectroscopy) experiments where necessary. 13C NMR spectra were assigned (aromatic C, CH, CH2, CH3) with the aid of DEPT (Distortionless Enhancement by Polarisation Transfer) experiments run in DEPT-90, DEPT-135 and DEPT-q modes. Specific assignments were made using HSQC (Heteronuclear Single Quantum Correlation) and HMBC (Heteronuclear Multiple Bond Correlation) experiments. All spectroscopic data for known compounds was in agreement with those previously reported unless otherwise stated. Samples used for comparison (stacked plots) were run at equal concentrations (6-8 mg per 0.65 mL solvent). Carbon analysis is given for compounds that are novel or where full analysis has not been published in the literature.
Infrared spectra were recorded on a Bruker Tensor 37 FT-IR spectrophotometer interfaced with Opus version 7.2.139.1294 over a range of 400–4000 cm−1. An average of 16 scans was taken for each spectrum obtained with a resolution of 4 cm-1. Melting points were measured on a Uni-Melt Thomas Hoover capillary melting point apparatus and are uncorrected. Melting points or boiling points were not obtained for semi-solids or oils.
Thin Layer Chromatography (TLC) was carried out on precoated silica gel plates (Merck 60 F254). Visualisation was achieved by UV light detection (254 nm or 366 nm), Wet flash chromatography was carried out using Kieselgel silica gel 60, 0.040–0.063 mm (Merck).
Low resolution mass spectra were recorded on a Waters Quattro Micro triple quadrupole spectrometer (QAA1202) in electron spray ionisation mode (ESI) using acetonitrile:water (1:1) containing 0.1% Formic acid as eluent. High resolution mass spectrometry (HRMS) spectra were recorded on Waters Vion IMS (model no. SAA055K) in electron spray ionisation mode (ESI) using acetonitrile:water (1:1) containing 0.1% Formic acid as eluent.
4.2. 6-Ethyl-9-Formyl-2,5,11-Trimethyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Iodide 20
Iodomethane (0.034 mL, 0.078 g, 0.552 mmol) was added to a stirred suspension of 6-ethyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 18 (0.152 g, 0.501 mmol) in anhydrous DMF (3.5 mL) and stirred at room temperature for 18 h. The reaction mixture was cooled on ice and cold diethyl ether (3 mL) was added. The resulting precipitate was collected by vacuum filtration and washed with hexane (3 mL) and diethyl ether (5 mL) to give the product as a yellow solid which was dried at 0.2 mbar (0.175 g, 78.8%). m.p. > 300 °C; vmax/cm−1 (KBr): 3072, 3047, 2934, 1672, 1573, 1242, 836; δH (300 MHz, DMSO-d6): 1.46 [3H, t, 7.0, N(6)CH2CH3], 3.09 [3H, s, C(5)CH3], 3.31 [3H, s C(11)CH3], 4.49 [3H, s, N(2)CH3], 4.80 [2H, q, J 7.0, N(6)CH2CH3], 7.97 [1H, d, J 8.6, C(7)H], 8.19 [1H, d, J 8.6, C(8)H], 8.52 [1H, d, J 7.3, C(4)H], 8.67 [1H, d, J 7.3, C(3)H], 8.91 [1H, s, C(10)H], 10.13 [1H, s, C(1)H], 10.16 [1H, s, C(9)CHO]; δC (150.9 MHz, DMSO-d6): 13.8 [CH3, C(5)CH3], 15.70 [CH3, N(6)CH2CH3], 15.74 [CH3, C(11)CH3], 41.1 [CH2, N(6)CH2CH3], 47.0 [CH3, N(2)CH3], 111.0 (CH, aromatic CH), 112.3 (C, aromatic C), 121.3 (C, aromatic C), 121.4 (CH, aromatic CH), 122.3 (C, aromatic C), 126.6 (C, aromatic C), 127.9 (CH, aromatic CH), 129.8 (CH, aromatic CH), 130.4 (C, aromatic C), 133.2 (CH, aromatic CH), 134.1 (C, aromatic C), 134.7 (C, aromatic C), 144.2 (C, aromatic C), 147.8 C(aromatic C), 148.0 (CH, aromatic CH), 192.6 [C, C(9)CHO]; m/z (ESI+): 317 [(M+H)+, 100%]; HRMS (ESI+): Exact mass calculated for C21H21N2O+ 317.1654. Found 317.1662.
4.3. 2,6-Diethyl-9-Formyl-5,11-Dimethyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Iodide 21
Iodoethane (0.044 mL, 0.085 g, 0.546 mmol) was added to a stirred solution of 6-ethyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 18 (0.150 g, 0.496 mmol) in anhydrous DMF (3.5 mL) and stirred at room temperature for 18 h. The reaction mixture was cooled on ice and cold diethyl ether (3 mL) was added. The resulting precipitate was collected by vacuum filtration and washed with hexane (3 mL) and diethyl ether (5 mL) to give the product as a yellow solid which was dried at 0.2 mbar (0.144 g, 63.5%). m.p. > 300 °C; vmax/cm−1 (KBr): 3046, 3014, 2973, 1671, 1587, 1574, 1242, 839; δH (600 MHz, DMSO-d6): 1.48 [3H, t, J 7.2, N(6)CH2CH3], 1.67 [3H, t, J 7.2, N(2)CH2CH3], 3.10 [3H, s, C(5)CH3], 3.34 [3H, s, C(11)CH3], 4.80 [4H, q, J 7.2, N(2)CH2CH3 and N(6)CH2CH3], 7.96 [1H, d, J 8.6, C(7)H], 8.18 [1H, dd, J 8.6, 1.5, C(8)H], 8.69 [2H, d, J 7.5, C(3)H, C(4)H], 8.92 [1H, s, C(10)H], 10.15 [1H, s, C(9)CHO], 10.20 [1H, s, C(1)H]; δC (150.9 MHz, DMSO-d6): 13.5 [CH3, C(5)CH3], 15.39 [CH3, N(6)CH2CH3], 15.43 [CH3, C(11)CH3], 16.7 [CH3, N(2)CH2CH3], 41.2 [CH2, N(6)CH2CH3], 56.0 [CH2, N(2)CH2CH3], 110.8 (CH, aromatic CH), 112.3 (C, aromatic C), 121.4 (C, aromatic C), 121.8 (CH, aromatic CH), 122.2 (C, aromatic C), 126.5 (C, aromatic C), 127.4 (CH, aromatic CH), 130.0 (C, aromatic C), 130.2 (CH, aromatic CH), 131.5 (CH, aromatic CH), 134.2 (C, aromatic C), 134.9 (C, aromatic C), 144.2 (C, aromatic C), 146.3 (CH, aromatic CH), 147.7 (C, aromatic C), 193.2 [C, C(9)CHO]; m/z (ESI+): 331 [(M+H)+, 100%]; HRMS (ESI+): Exact mass calculated for C22H23N2O+ 331.1810. Found 331.1804.
4.4. 6-Ethyl-9-Formyl-2-Isopropyl-5,11-Dimethyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Iodide 22
2-Iodopropane (0.055 mL, 0.093 g, 0.546 mmol) was added to a stirred solution of 6-ethyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 18 (0.150 g, 0.496 mmol) in 1,4-dioxane (8 mL) and stirred at reflux for 16 h. Starting material was still present when the reaction mixture was examined using TLC. Additional 2-iodopropane (0.10 mL, 0.169 g, 0.992 mmol) was added and heated to reflux for a further 24 h. The mixture was cooled on ice and cold diethyl ether (3 mL) was added. The resultant precipitate was filtered and washed with hexane (3 mL) and diethyl ether (5 mL) to give the crude product. Recrystallisation from methanol gave the desired product as a yellow solid, which was dried at 0.2 mbar (0.127 g, 54.4%). m.p. > 300 °C; vmax/cm−1 (KBr): 2977, 2728, 1669, 1586, 1404, 1243, 1098; δH (300 MHz, DMSO-d6): 1.48 [3H, t, J 7.2, N(6)CH2CH3], 1.76 [6H, d, J 6.8, N(2)CH(CH3)2], 3.13 [3H, s, C(5)CH3], 3.41 [3H, s, C(11)CH3], 4.84 [2H, q, J 7.2, N(6)CH2CH3], 5.30 [1H, sept, J 6.8, N(2)CH(CH3)2], 8.00 [1H, d, J 8.6, C(7)H], 8.20 [1H, d, J 8.6, C(8)H], 8.72 [1H, d, J 7.5, C(4)H], 8.80 [1H, d, J 7.5, C(3)H], 8.99 [1H, s, C(10)H], 10.17 [1H, s, C(9)CHO], 10.19 [1H, s, C(1)H]; δC (150.9 MHz, DMSO-d6): 13.5 [CH3, C(5)CH3], 15.38 [CH3, C(11)CH3], 15.41 [CH3, N(6)CH2CH3], 22.7 [2 x CH3, N(2)CH(CH3)2], 41.1 [CH2, N(6)CH2CH3], 63.7 [CH, N(2)CH(CH3)2] 110.7 (CH, aromatic CH), 112.3 (C, aromatic C), 121.3 (C, aromatic C), 122.1 (CH, aromatic CH), 122.2 (C, aromatic C), 126.6 (C, aromatic C), 127.4 (CH, aromatic CH), 129.0 (CH, aromatic CH), 129.9 (C, aromatic C), 130.2 (CH, aromatic CH), 134.3 (C, aromatic C), 135.3 (C, aromatic C), 144.2 (C, aromatic C), 145.2 (CH, aromatic CH), 147.8 (C, aromatic C), 193.2 [C, C(9)CHO]; m/z (ESI+): 345 [(M+H)+, 100%]; HRMS (ESI+): Exact mass calculated for C23H25N2O+ 345.1967. Found 345.1958.
4.5. 2-(5-Cyanopentyl)-6-Ethyl-9-Formyl-5,11-Dimethyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Bromide 23
6-Bromohexanenitrile (0.052 mL, 69.7 mg, 0.396 mmol) was added to a stirred suspension of 6-ethyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 18 (100 mg, 0.331 mmol) in dimethylformamide (5 mL) and heated to 120 °C for four hours. The reaction mixture was cooled on ice and cold diethyl ether (5 mL) added. The resultant precipitate was collected by filtration and washed with hexane to give the product as a yellow solid which was dried at 0.2 mbar (123 mg, 77.9%). m.p. 246–248 °C; νmax/cm−1 (KBr): 3374, 2932, 2240, 1680, 1642, 1580, 1460, 1395, 1357, 1239, 814; δH (500 MHz, DMSO-d6): 1.40 – 1.52 [5H, m, N(6)CH2CH3, C(3′)H2], 1.62 – 1.71 [2H, m, C(4′)H2], 2.03 – 2.11 [2H, m, C(2′)H2], 2.55 [2H, t, J 7.0, C(5′)H2], 3.09 [3H, s, C(5)CH3], 3.34 [3H, s, C(11)CH3], 4.75 – 4.83 [4H, m, N(6)CH2CH3, C(1′)H2], 7.96 [1H, d, J 8.6, C(7)H], 8.17 [1H, d, J 8.4, C(8)H], 8.66 [1H, d, J 7.2, C(4)H], 8.71 [1H, d, J 7.2, C(3)H], 8.91 [1H, s, C(10)H], 10.14 [1H, s, C(9)CHO], 10.23 [1H, s, C(1)H]; δC (125.8 MHz, DMSO-d6): 13.8 [CH3, C(5)CH3], 15.7 [CH3, N(6)CH2CH3], 15.9 [CH3, C(11)CH3], 16.5 [CH2, C(5′)H2], 24.7 [CH2, C(4′)H2], 25.2 [CH2, C(3′)H2], 30.5 [CH2, C(2′)H2], 41.1 [CH2, N(6)CH2CH3], 59.7 [CH2, C(1′)H2], 111.1 (CH, aromatic CH), 112.4 (C, aromatic C), 121.1 (C, CN), 121.5 (C, aromatic C), 121.8 (CH, aromatic CH), 122.3 (C, aromatic C), 126.8 (C, aromatic C), 127.9 (CH, aromatic CH), 129.8 (CH, aromatic CH), 130.5 (C, aromatic C), 132.2 (CH, aromatic CH), 134.5 (C, aromatic C), 134.9 (C, aromatic C), 144.3 (C, aromatic C), 147.3 (CH, aromatic CH), 147.8 (C, aromatic C), 192.6 [C, C(9)CHO]; m/z (ESI+): 398 [(M)+, 100%]; HRMS (ESI+): Exact mass calculated for C26H28N3O+ 398.2232. Found 398.2237.
4.6. 6-Ethyl-9-Formyl-5,11-Dimethyl-2-Tetradecyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Bromide 24
1-Bromotetradecane (0.13 mL, 0.120g, 0.434 mmol) was added to a stirred solution of 6-ethyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 18 (0.119g, 0.394 mmol) in anhydrous DMF (8 mL) and stirred at reflux for 16 h. The mixture was cooled on ice and cold diethyl ether was added (3 mL). The resultant precipitate was filtered and washed with hexane (3 mL) and diethyl ether (5 mL) to give the product as a yellow solid which was dried at 0.2 mbar (0.156 g, 68.5%). m.p. 194–197 °C; vmax/cm−1 (KBr): 2922, 2852, 1674, 1586, 1356, 1243, 1101, 810; δH (300 MHz, DMSO-d6): 0.83 [3H, t, J 6.6, C(14′)H], 1.12-1.41 [22H, m, (3′)H2-C(13′)H2], 1.48 [3H, t, 7.1, N(6)CH2CH3], 1.96-2.10 [2H, m, C(2′)H], 3.11 [3H, s, C(5)CH3], 3.37 [1H, s, C(11)CH3], 4.76 [2H, t, J 7.5, C(1′)H], 4.83 [2H, q, J 7.1, N(6)CH2CH3], 7.99 [1H, d, J 8.6, C(7)H], 8.20 [1H, dd, J 8.6, 1.4, C(8)H], 8.66 [1H, d, J 7.6, C(4)H], 8.72 [1H, d, J 7.6, C(3)H], 8.96 [1H, d, J 1.2, C(10)H], 10.17 [1H, s, C(9)CHO], 10.24 [1H, s, C(1)H]; δC (150.9 MHz, DMSO-d6): 13.6 [CH3, C(5)CH3], 14.4 [CH3, C(14′)H3], 15.7 [CH3, N(6)CH2CH3], 15.9 [CH3, C(11)CH3], 22.5 (CH2), 26.1 (CH2), 29.0 (CH2), 29.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (2 x CH2), 29.5 (2 x CH2), 31.4 (CH2), 31.7 [CH2, C(2′)H2], 41.0 [CH2, N(6)CH2CH3], 59.9 [CH2, C(1′)H2], 101.7 (CH, aromatic CH), 112.2 (C, aromatic C), 121.3 (C, aromatic C), 121.7 (CH, aromatic CH), 122.1 (C, aromatic C), 126.5 (C, aromatic C), 127.7 (CH, aromatic CH), 129.6 (CH, aromatic CH), 130.3 (C, aromatic C), 132.1 (CH, aromatic CH), 134.2 (C, aromatic C), 134.7 (C, aromatic C), 144.0 (C, aromatic C), 147.0 (CH, aromatic CH), 147.5 (C, aromatic C), 192.4 [C, C(9)CHO]; m/z (ESI+): 499 [(M+H)+, 100%]; HRMS (ESI+): Exact mass calculated for C34H47N2O+ 499.3688. Found 499.3691.
4.7. 6-Ethyl-9-Formyl-5,11-Dimethyl-2-Octadecyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Bromide 25
1-Bromooctadecane (140 mg, 0.42 mmol) was added to a stirred suspension of 6-ethyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 18 (105 mg, 0.35 mmol) in dimethylformamide (5 mL) and heated to 120 °C for four hours. The reaction mixture was cooled on ice and cold diethyl ether (5 mL) added. The resultant precipitate was collected by filtration and washed with hexane to give the product as a yellow solid which was dried at 0.2 mbar (182.5 mg, 82.0%). m.p. 199–203 °C; νmax/cm−1 (KBr): 3418, 2921, 2851, 1680, 1581, 1459, 1399, 1357, 1242, 1101, 810; δH (500 MHz, DMSO-d6): 0.83 [3H, t, J 6.6, C(18′)H3], 1.10–1.40 [30H, m, C(3′)H2–C(17′)H2], 1.48 [3H, t, J 6.8, N(6)CH2CH3], 1.99–2.08 [2H, m, C(2′)H2], 3.11 [3H, s, C(5)CH3], 3.37 [3H, s, C(11)CH3], 4.76 [2H, t, J 6.8, C(1′)H2], 4.82 [2H, q, J 6.7, N(6)CH2CH3], 7.99 [1H, d, J 8.5, C(7)H], 8.20 [1H, d, J 8.4, C(8)H], 8.66 [1H, d, J 7.1, C(4)H], 8.72 [1H, d, J 7.0, C(3)H], 8.96 [1H, s, C(10)H], 10.16 [1H, s, C(9)CHO], 10.23 [1H, s, C(1)H]; δC (125.8 MHz, DMSO-d6): 13.8 [CH3, C(5)CH3], 14.4 [CH3, C(18′)H3], 15.7 [CH3, N(6)CH2CH3], 15.9 [CH3, C(11)CH3], 22.5 (CH2), 26.1 (CH2), 28.9 (CH2), 29.1 (CH2), 29.26 (CH2), 29.32 (CH2), 29.5 (8 × CH2), 31.3 [CH2, C(2′)H2], 31.7 (CH2), 41.1 [CH2, N(6)CH2CH3], 60.1 [CH2, C(1′)H2], 111.1 (CH, aromatic CH), 112.5 (C, aromatic C), 121.5 (C, aromatic C), 121.8 (CH, aromatic CH), 122.4 (C, aromatic C), 126.8 (C, aromatic C), 127.9 (CH, aromatic CH), 129.9 (CH, aromatic CH), 130.6 (C, aromatic C), 132.2 (CH, aromatic CH), 134.5 (C, aromatic C), 135.0 (C, aromatic C), 144.4 (C, aromatic C), 147.3 (CH, aromatic CH), 147.8 (C, aromatic C), 192.6 [C, C(9)CHO]; m/z (ESI+): 555 [(M)+, 100%]; HRMS (ESI+): Exact mass calculated for C38H55N2O+ 555.4314. Found 555.4293.
4.8. 2-Ethyl-9-Formyl-6-Isopropyl-5,11-Dimethyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Iodide 26
Iodoethane (0.05 mL, 97 mg, 0.62 mmol) was added to a suspension of 6-isopropyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 19 (99 mg, 0.31 mmol) in dimethylformamide (5 mL) and stirred at room temperature for 16 h. Thin layer chromatography analysis indicated a large amount of starting material was still present. Additional iodoethane (0.05 mL, 97 mg, 0.62 mmol) was added and the reaction was stirred at room temperature for a further 24 h. The mixture was cooled on ice and diethyl ether (5 mL) added. The resultant precipitate was filtered, washed with hexane and dried by heating under reduced pressure (110 °C, 0.2 mbar) to give the product as a yellow solid (116.1 mg, 78.8%). m.p. > 300 °C without melting; νmax/cm−1 (KBr): 2974, 1675, 1579, 1451, 1397, 1310, 1246, 1209, 1100, 807; δH (300 MHz, DMSO-d6): 1.65 [3H, t, J 7.2, N(2)CH2CH3], 1.73 [6H, d, J 7.0, N(6)CH(CH3)2], 3.08 [3H, s, C(5)CH3], 3.40 [3H, s, C(11)CH3], 4.81 [2H, q, J 7.2, N(2)CH2CH3], 5.61 [1H, sept, J 6.8, N(6)CH(CH3)2], 8.14–8.19 [2H, m, C(7)H, C(8)H], 8.65 – 8.72 [2H, m, C(3)H, C(4)H], 9.02 [1H, s, C(10)H], 10.20 [1H, s, C(9)CHO], 10.24 [1H, s, C(1)H]; δC (150.9 MHz, DMSO-d6): 15.7 [CH3, C(5)CH3], 15.8 [CH3, C(11)CH3], 17.1 [CH3, N(2)CH2CH3], 21.3 [2 × CH3, N(6)CH(CH3)2], 50.4 [CH, N(6)CH(CH3)2], 55.8 [CH2, N(2)CH2CH3], 112.8 (C, aromatic C), 114.6 (CH, aromatic CH), 121.7 (C, aromatic C), 122.2 (CH, aromatic CH), 124.0 (C, aromatic C), 126.9 (C, aromatic C), 128.1 (CH, aromatic CH), 129.0 (CH, aromatic CH), 130.2 (C, aromatic C), 132.0 (CH, aromatic CH), 134.0 (C, aromatic C), 135.5 (C, aromatic C), 146.4 (C, aromatic C), 146.8 (C, aromatic C), 147.2 (CH, aromatic CH), 192.8 [C, C(9)CHO]; m/z (ESI+): 345 [(M)+, 100%]; HRMS (ESI+): Exact mass calculated for C23H25N2O+ 345.1967. Found 345.1965.
4.9. 2-(5-Cyanopentyl)-9-Formyl-6-Isopropyl-5,11-Dimethyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Bromide 27
6-Bromohexanenitrile (0.04 mL, 0.049g, 0.278 mmol) was added to a stirred suspension of 6-isopropyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 19 (0.080 g, 0.253 mmol) in anhydrous DMF (3 mL) and stirred at 120 °C for 16 h. TLC showed the presence of starting material so a further portion of 6-bromohexanenitrile (0.018 mL, 0.024 g, 0.139 mmol) was added and the reaction was heated to 120 °C for a further 24 h. The reaction mixture was cooled on ice and cold diethyl ether was added (3 mL). The resultant precipitate was filtered and washed with hexane (3 mL) and diethyl ether (5 mL) to give the product as a yellow solid which was dried at 0.2 mbar (0.089 g, 71.9%). m.p. 253–255 °C; vmax/cm−1 (KBr): 3383, 2937, 2241, 1677, 1585, 1399, 1251, 1101; δH (600 MHz, DMSO-d6): 1.42-1.51 [2H, m, C(3′)H2], 1.67 [2H, quin, J 7.4, C(4′)H2], 1.73 [6H, d, J 6.9, N(6)CH(CH3)2], 2.08 [2H, quin, J 7.4, C(2′)H2], 2.55 [2H, t, J 7.4, C(5′)H2], 3.08 [3H, s, C(5)CH3], 3.40 [3H, s, C(11)CH3], 4.80 [2H, t, J 7.4, C(1′)H2], 5.61 [1H, sept, J 6.9, N(6)CH(CH3)2], 8.17 [2H, s, C(7)H, C(8)H], 8.71 [2H, s, C(3)H, C(4)H], 9.02 [1H, s, C(10)H], 10.20 [1H, s, C(9)CHO], 10.28 [1H, s, C(1)H]; δC (150.9 MHz, DMSO-d6): 15.7 [CH3, C(5)CH3], 15.9 [CH3, C(11)CH3], 16.5 [CH2, C(5′)H2], 21.3 [2 x CH3, N(6)CH(CH3)2], 24.7 [CH2, C(4′)H2], 25.1 [CH2, C(3′)H2], 30.5 [CH2, C(2′)H2], 50.4 [CH, N(6)CH(CH3)2], 59.7 [CH2, C(1′)H2], 112.8 (C, aromatic C), 114.6 (CH, aromatic CH, one of C(7)H or C(8)H), 121.1 (C, CN), 121.8 (C, aromatic C), 122.0 [CH, C(1)H], 124.1 (C, aromatic C), 127.0 (C, aromatic C), 128.1 (CH, aromatic CH), 129.1 (CH, aromatic CH, one of C(7)H or C(8)H), 130.3 (C, aromatic C), 132.3 [CH, C(3)H], 134.1 (C, aromatic C), 135.6 (C, aromatic C), 146.5 (C, aromatic C), 146.8 (C, aromatic C), 147.4 (CH, aromatic CH), 192.7 [C(9)CHO]; m/z (ESI+): 412 [(M+H)+, 100%]; HRMS (ESI+): Exact mass calculated for C27H30N3O+ 412.2389. Found 412.2393.
4.10. 9-Formyl-6-Isopropyl-5,11-Dimethyl-2-Tetradecyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Bromide 28
1-Bromotetradecane (0.11 mL, 102 mg, 0.368 mmol) was added to a stirred suspension of of 6-isopropyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 19 (100 mg, 0.316 mmol) and heated to 120 °C for 4 h. Following thin layer chromatography analysis, further 1-bromotetradecane (0.06 mL, 56 mg, 0.201 mmol) was added and the reaction was heated to 120 °C for 16 h. The reaction was cooled on ice and cold diethyl ether (5 mL) added. The resultant precipitate was filtered, washed with hexane and dried at 0.2 mbar to give the product as a yellow solid (149 mg, 79.7%). m.p. 225–227 °C; νmax/cm−1 (KBr): 3421, 2924, 2852, 1682, 1583, 1461, 1396, 1245, 1102; δH (500 MHz, DMSO-d6): 0.83 [3H, t, J 6.9, C(14′)H3], 1.12–1.39 [22H, m, C(3′)H2–C(13′)H2], 1.73 [6H, d, J 6.9, N(6)CH(CH3)2], 1.99–2.09 [2H, m, C(2′)H2], 3.07 [3H, s, C(5)CH3], 3.39 [3H, s, C(11)CH3], 4.78 [2H, t, J 7.3, C(1′)H2], 5.60 [1H, sept, J 6.7, N(6)CH(CH3)2], 8.16 [2H, br s, C(7)H, C(8)H], 8.69 [2H, br s, C(3)H, C(4)H], 9.00 [1H, s, C(10)H], 10.19 [1H, s, C(9)CHO], 10.26 [1H, s, C(1)H]; δC (125.8 MHz, DMSO-d6): 14.4 [CH3, C(14′)H3], 15.7 [CH3, C(5)CH3], 15.9 [CH3, C(11)CH3], 21.3 [2 × CH3, N(6)CH(CH3)2], 22.5 (CH2), 26.0 (CH2), 28.9 (CH2), 29.1 (CH2), 29.27 (CH2), 29.33 (CH2), 29.42 (CH2), 29.44 (CH2), 29.5 (2 × CH2), 31.3 [CH2, C(2′)H2], 31.7 (CH2), 50.4 [CH, N(6)CH(CH3)2], 60.2 [CH2, C(1′)H2], 112.8 (C, aromatic C), 114.6 (CH, aromatic CH), 121.8 (C, aromatic C), 122.1 (CH, aromatic CH), 124.1 (C, aromatic C), 127.0 (C, aromatic C), 128.0 (CH, aromatic CH), 129.1 (CH, aromatic CH), 130.3 (C, aromatic C), 132.3 (CH, aromatic CH), 134.0 (C, aromatic C), 135.5 (C, aromatic C), 146.4 (C, aromatic C), 146.8 (C, aromatic C), 147.3 (CH, aromatic CH), 192.7 [C, C(9)CHO]; m/z (ESI+): 513 [(M)+, 100%]; HRMS (ESI+): Exact mass calculated for C35H49N2O+ 513.3845. Found 513.3844.
4.11. 9-Formyl-6-Isopropyl-5,11-Dimethyl-2-Octadecyl-6H-Pyrido[4,3-b]Carbazol-2-Ium Bromide 29
1-Bromooctadecane (0.10 mL, 0.094 g, 0.278 mmol) was added to a stirred suspension of 6-isopropyl-5,11-dimethyl-6H-pyrido[4,3-b]carbazole-9-carbaldehyde 19 (0.080 g, 0.253 mmol) in anhydrous DMF (3 mL) and stirred at 120 °C for 16 h. A red-brown solution formed on heating. The reaction mixture was cooled on ice and cold diethyl ether was added (3 mL). The resultant precipitate was filtered and washed with hexane (3 mL) and diethyl ether (5 mL) to give the product as a yellow solid which was dried at 0.2 mbar (0.128 g, 78.1%). m.p. 138-141 °C; vmax/cm−1 (KBr): 2953, 2919, 2850, 1675, 1592, 1251, 1101; δH (600 MHz, DMSO-d6): 0.83 [3H, t, J 7.1, C(18′)H3], 1.11–1.39 [30H, m, C(3′)H2-C(17′)H2], 1.73 [6H, d, J 7.0, N(6)CH(CH3)2], 2.03 [2H, quin, J 7.1, C(2′)H2], 3.08 [3H, s, C(5)CH3], 3.39 [3H, s, C(11)CH3], 4.78 [2H, t, J 7.5, C(1′)H2], 5.60 [1H, sept, J 7.0, N(6)CH(CH3)2], 8.16 [2H, s, C(7)H, C(8)H], 8.69 [2H, s, C(3)H, C(4)H], 8.99 [1H, s, C(10)H], 10.18 [1H, s, C(9)CHO], 10.26 [1H, s, C(1)H]; δC (150.9 MHz, DMSO-d6): 14.4 [CH3, C(18′)H3], 15.7 [CH3, C(5)CH3], 15.9 [CH3, C(11)CH3], 21.3 [2 x CH3, N(6)CH(CH3)2], 22.5 (CH2), 26.0 (CH2), 28.9 (CH2), 29.1 (CH2), 29.24 (CH2), 29.30 (CH2), 29.4 (CH2), 29.5 (6 x CH2), 31.3 [CH2, C(2′)H2], 31.7 (CH2), 40.4 (CH2), 50.4 [CH, N(6)CH(CH3)2], 60.2 [CH2, C(1′)H2], 112.8 (C, aromatic C), 114.7 (CH, aromatic CH), 121.9 (C, aromatic C), 122.1 (CH, aromatic CH), 124.1 (C, aromatic C), 127.1 (C, aromatic C), 128.1 (CH, aromatic CH), 129.2 (CH, aromatic CH), 130.3 (C, aromatic C), 132.3 (CH, aromatic CH), 134.1 (C, aromatic C), 135.6 (C, aromatic C), 146.5 (C, aromatic C), 146.9 (C, aromatic C), 147.3 (CH, aromatic CH), 192.7 [C, C(9)CHO]; m/z (ESI+): 596 [(M+H)+, 100%]; HRMS (ESI+): Exact mass calculated for C39H57N2O+ 569.4471. Found 569.4462.
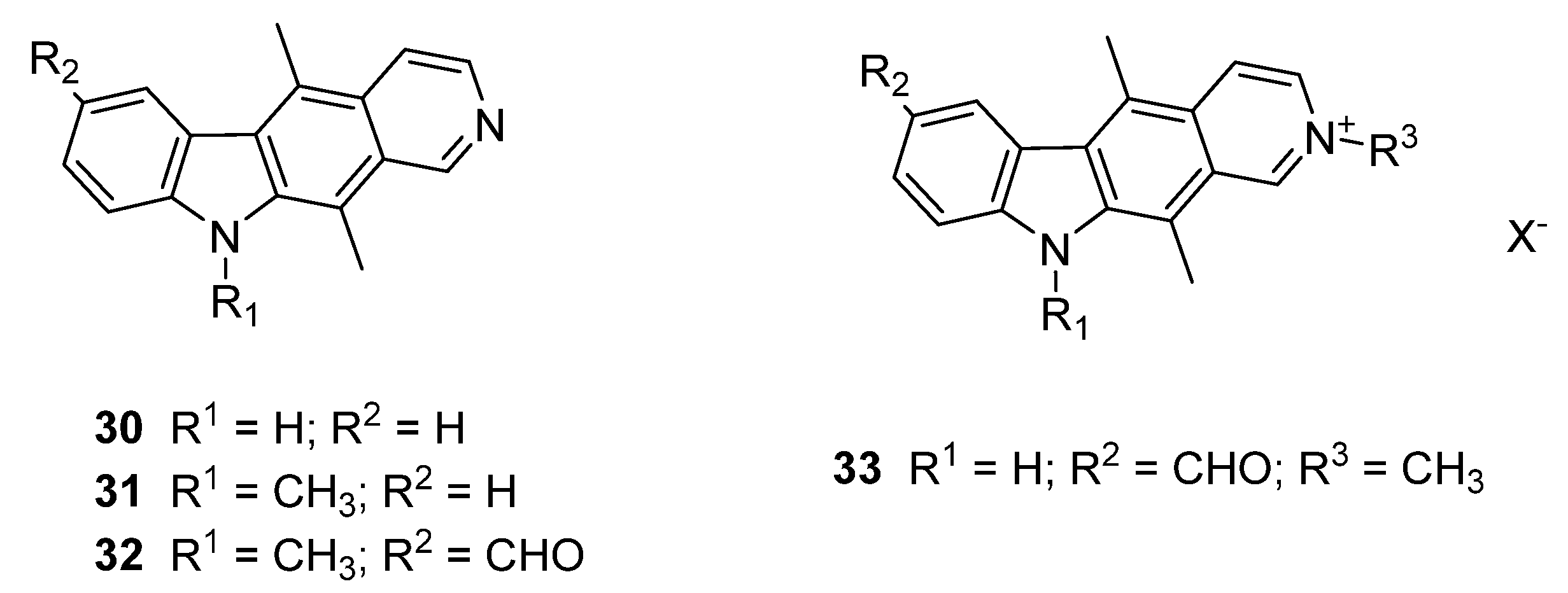
4.12. 5,10,11-Trimethyl-10H-Pyrido[3,4-b]Carbazole 31
Sodium hydride (134 mg, 60% dispersion in mineral oil, equivalent to 80 mg sodium hydride, 3.3 mmol) was suspended in dimethylformamide (5 mL) under nitrogen and 5,11-dimethyl-10
H-pyrido[3,4-
b]carbazole
30 (274 mg, 1.11 mmol) added portionwise, forming a deep red suspension which was stirred at room temperature for 45 min. Iodomethane (0.07 mL, 160 mg, 1.12 mmol) in dimethylformamide (3 mL) was added dropwise and the reaction was stirred overnight. The reaction mixture was cooled on ice and water (30 mL) added. The resultant yellow precipitate was filtered and washed with excess water to give the product which was used without further purification (276 mg, 95.5%). m.p. 165–167 °C (Lit. m.p. 157–159) [
45]; ν
max/cm
−1 (KBr): 3047, 3016, 2926, 1591, 1481, 1388, 1356, 1329, 1304, 1234, 989, 741; δ
H (300 MHz, DMSO-
d6): 3.02 [3H, s, C(11)C
H3], 3.13 [3H, s, C(5)C
H3], 4.09 [3H, s, N(10)C
H3], 7.23–7.33 [1H, m, C(7)H], 7.56–7.64 [2H, m, C(8)H, C(9)H], 8.07 [1H, d,
J 6.0, C(4)H], 8.34 [1H, d,
J 7.9, C(6)H], 8.42 [1H, d,
J 6.0, C(3)H], 9.63 [1H, s, C(1)H]; m/z (ESI
+): 261 [(M+H)
+, 100%].
4.13. 5,10,11-Trimethyl-10H-Pyrido[3,4-b]Carbazole-7-Carbaldehyde 32
5,10,11-Trimethyl-10H-pyrido[3,4-b]carbazole 31 (920 mg, 3.53 mmol) was stirred in trifluoroacetic acid (50 mL) and hexamethylenetetramine (4.95 g, 35.3 mmol) added portionwise. The solution was refluxed for 30 min, cooled to room temperature and concentrated to one third volume under reduced pressure. Water (250 mL) was added and pH was adjusted to 6 with sodium bicarbonate on ice (Caution: vigorous reaction). Dichloromethane – methanol 90:10 (1 × 200mL) was added and an emulsion formed which resolved upon stirring overnight. The organic layer was separated and the aqueous layer extracted with further dichloromethane – methanol 90:10 (2 × 100 mL). Combined organic extracts were washed with water (2 × 100 mL) and brine (1 × 100 mL), dried over magnesium sulfate and solvent removed under reduced pressure to give the product as a yellow solid (916 mg, 89.8%). m.p. 169–171 °C; vmax/cm−1 (KBr):3417, 2924, 2855, 1678, 1585, 1461, 1360, 1303, 1203, 1097, 802; δH (500 MHz, DMSO-d6): 2.79 [3H, s, C(11)CH3], 2.94 [3H, s, C(5)CH3], 3.93 [3H, s, N(10)CH3], 7.57 [1H, d, J 8.5, C(9)H], 7.94 [1H, d, J 5.9, C(4)H], 7.98 [1H, d, J 8.5, C(8)H], 8.40 [1H, d, J 5.7, C(3)H], 8.50 [1H, s, C(6)H], 9.53 [1H, s, C(1)H], 9.98 [1H, s, C(7)CHO]; δC (125.8 MHz, DMSO-d6): 13.3 [CH3, C(5)CH3], 14.8 [CH3, C(11)CH3], 34.3 [CH3, N(10)CH3], 109.5 (CH, aromatic CH), 113.0 (C, aromatic C), 116.8 (CH, aromatic CH), 122.3 (C, aromatic C), 125.7 (C, aromatic C), 126.3 (C, aromatic C), 126.7 (C, aromatic C), 127.3 (CH, aromatic CH), 128.7 (C, aromatic C), 128.9 (CH, aromatic CH), 129.4 (C, aromatic C), 139.1 (C, aromatic C), 139.8 (CH, aromatic CH), 148.7 (C, aromatic C), 149.3 (CH, aromatic CH), 194.1 [C, C(7)CHO]; m/z (ESI+): 289 [(M + H)+,100%]; HRMS (ESI+): Exact mass calculated for C19H17N2O+ 289.1341. Found 289.1341.
4.14. 7-Formyl-2,5,11-Trimethyl-10H-Pyrido[3,4-b]Carbazol-2-Ium Iodide 33
5,11-Dimethyl-10
H-pyrido[3,4-
b]carbazole
30 (1.72 g, 6.98 mmol) was dissolved in trifluoroacetic acid (100 mL) and hexamethylenetetramine (9.79 g, 69.8 mmol) gradually added over five minutes. The mixture was heated to reflux for 40 min and upon cooling, concentrated to half volume under reduced pressure. The mixture was placed on ice and water (200 mL) added. The solution was neutralised with solid sodium bicarbonate (Caution: vigorous reaction) and the resultant orange precipitate isolated by filtration. Column chromatography on neutral alumina eluting with dichloromethane–methanol 95:5 gave the product 5,11-Dimethyl-10
H-pyrido[3,4-
b]carbazole-7-carbaldehyde (1.38 g, 72.3%). m.p. 294–296 °C (Lit. m.p. 270–271 °C)[
45]; ν
max/cm
−1 (KBr): 3029, 2864, 1676, 1600, 1471, 1400, 1380, 1317, 1290, 1217, 1202, 1113, 1015, 807; δ
H (300 MHz, DMSO-
d6): 2.94 [3H, s, C(11)C
H3], 3.16 [3H, s, C(5)C
H3], 7.66 [1H, d,
J 8.4, C(9)H], 8.06 [1H, dd,
J 8.4, 1.3, C(8)H], 8.14 [1H, d,
J 6.0, C(4)H], 8.46 [1H, d,
J 6.1, C(3)H], 8.86 [1H, s, C(6)H], 9.59 [1H, br s, C(1)H], 10.09 [1H, s, C(7)C
HO], 11.90 [1H, s, N(10)H]; m/z (ESI
+): 275 [(M + H)
+, 100%].
5,11-Dimethyl-10
H-pyrido[3,4-
b]carbazole-7-carbaldehyde (188 mg, 0.68 mmol) was then suspended in dimethylformamide (5 mL) and iodomethane (0.06 mL, 136 mg, 0.96 mmol) added. The mixture was stirred for 16 h at room temperature, cooled on ice and cold diethyl ether (5 mL) added. The resultant precipitate was filtered under nitrogen, washed with hexane and dried under vacuum (0.2 mbar) to give the product as a red solid (254 mg, 89.7%). m.p. > 300 °C without melting (Lit. m.p. > 300 °C without melting) [
45]; ν
max/cm
−1 (KBr): 3418, 3142, 3008, 2838, 2749, 1677, 1638, 1621, 1600, 1467, 1408, 1385, 1318, 1303, 1241, 1199, 1113; δ
H (300 MHz, DMSO-
d6): 2.98 [3H, s, C(11)C
H3], 3.18 [3H, s, C(5)C
H3], 4.54 [3H, s, N(2)C
H3], 7.69 [1H, d,
J 8.5, C(9)H], 8.11 [1H, dd,
J 8.5, 0.9, C(8)H], 8.49 [1H, d,
J 7.0, C(4)H], 8.75 [1H, d,
J 7.0, C(3)H], 8.86 [1H, s, C(6)H], 9.99 [1H, s, C(1)H], 10.11 [1H, s, C(7)C
HO], 12.34 [1H, s, N(10)H]; m/z (ESI
+): 289 [(M)
+, 100%].
4.15. Phytophthora infestans Mycelial Growth Assays
Phytophthora infestans stain 88069 (mating type A1) was originally obtained from Professor Sophien Kamoun (The Sainsbury Laboratory, Norwich, UK) and was cultured and maintained on RyeA agar at 20 °C in the dark [
46]. For mycelial growth assay, compounds were dissolved in DMSO in a vial and 15mL Rye B agar added, at a temperature of approximately 50 °C (the final concentration of DMSO was 0.1%). The vial was shaken to distribute the contents and the mixture was then poured into a clean, sterile 10 cm petri dish. A 10 mm plug of
P. infestans mycelium was placed in the centre of the dish, which was then sealed with parafilm. The experiments were carried out in triplicate at 25 μM (unless otherwise stated), stored at 20 °C in the dark and checked periodically for mycelial growth.
4.16. Phytophthora infestans Zoosporogenesis and Zoospore Motility Assays
Zoosporogenesis was assayed as described by Lu et al. [
38]. Following two weeks of culture on Rye B agar, sporangia were collected from plates in 5 mL of modified Petri’s solution (MPS, 5 mM CaCl
2, 1 mM MgSO
4, 1 mM KH
2PO
4 and 0.8 mM KCl), by scraping with a scalpel blade and were transferred to a 50 mL sterile tube. Plates were washed with another 5 mL of MPS and this was combined with the first 5 mL. To initiate zoospore release, sporangia were placed on ice, in the dark, for 3h. Sporangia or zoospores were counted by brightfield microscopy using a 10× objective and a haemocytometer, in duplicate.
The motility of harvested zoospores was analysed essentially as described by Appiah et al. [
47]. In brief, test compounds were added to 100 μL of zoospores, which were immediately transferred to a 100 mm
2 square chamber on a microscope chamber, formed by extruding petroleum jelly from a 5 mL syringe. This chamber was sealed with a 0-thickness glass cover-slip, was inverted and placed on a 100× magnification, 1.3 numerical aperture oil-immersion objective of an Olympus IX51 microscope. Using brightfield illumination, the motion of the zoospores within this chamber were recorded for 10 s, at a rate of 10 frames/s, using a Hamamatsu ORCA ER CCD video camera ((Hamamatsu Photonics Ltd., Hertfordshire, UK) and Andor IQ 1.9 acquisition software (Belfast, Northern Ireland). Video recordings were exported as multi-dimensional TIFF files and zoospore trajectories were plotted using the Manual Tracking plugin of ImageJ (
https://imagej.nih.gov/ij/plugins/track/Manual%20Tracking%20plugin.pdf). Cell traces were exported in Microsoft Excel format and were analysed using the Chemotaxis and Migration Tool from Ibidi (
https://ibidi.com/img/cms/resources/AG/FL_AG_035_Chemotaxis_150dpi.pdf). For each zoospore, this generated numerical data on the velocity, accumulated distance travelled, Euclidean (straight-line from origin to furthest point) distance travelled and directionality (“straightness” of travel, where 1 represents completely linear travel and 0, completely circular).
4.17. XTT Reduction Assay
Human embryonic kidney-293T (HEK-293T) cells were obtained from LGC Standards (Middlesex, UK) and were maintained in Dulbecco’s Modified Eagles Medium containing 10% foetal bovine serum, 100 units/mL penicillin and 100 μg/mL streptomycin, at 37 °C in a humidified atmosphere of 5% CO
2/95% air. For assays, cells were subcultured on 96-well microtitre plates at a density of 5 × 10
4 cells/well, in a volume of 50 mL. Following overnight culture, 50 mL of each test compound at twice their final concentration (50 mM for all apart from cinnamaldehyde (2 mM) and Mancozeb (200 M)) was added and incubated for another 24 h. Cells were incubated with 100 mL XTT reagents (Sigma-Aldrich, Ireland) for an additional 4 h, after which, the absorbance of the extracellular formazan reduction product was measured at a wavelength of of 490 nm, with a reference wavelength of 675 nm [
41,
42], using a microtitre plate spectrophotometer.
4.18. Statistical Methods
Numerical data were statistically compared using one-way analysis of variance (ANOVA) and Tukey’s post-hoc test (using ezANOVA software, available at:
https://people.cas.sc.edu/rorden/ezanova/index.html.) A probability threshold of
p < 0.05 was taken as statistically significant. Generation of histograms and linear regression analyses were performed using Microsoft Excel. Concentration-growth inhibition data were analysed using GraphPad Prism software version 4.03 (San Diago, CA, USA).

 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}