A Genome-Wide Association Study Identifies Quantitative Trait Loci Affecting Hematological Traits in Camelus bactrianus


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Sample Collection
2.3. Genotyping
2.4. Sequence Analyses and Single-Nucleotide Polymorphism (SNP) Calling
2.5. Estimating the Linkage Disequilibrium (LD) and Deducing the Population Structure
2.6. Statistical Analyses
2.7. Genome-Wide Association Study
3. Results
3.1. Quality Assessment and Statistics of the Sequencing Data
3.2. Mapping to the Reference Genome
3.3. SNP Calling and Annotation
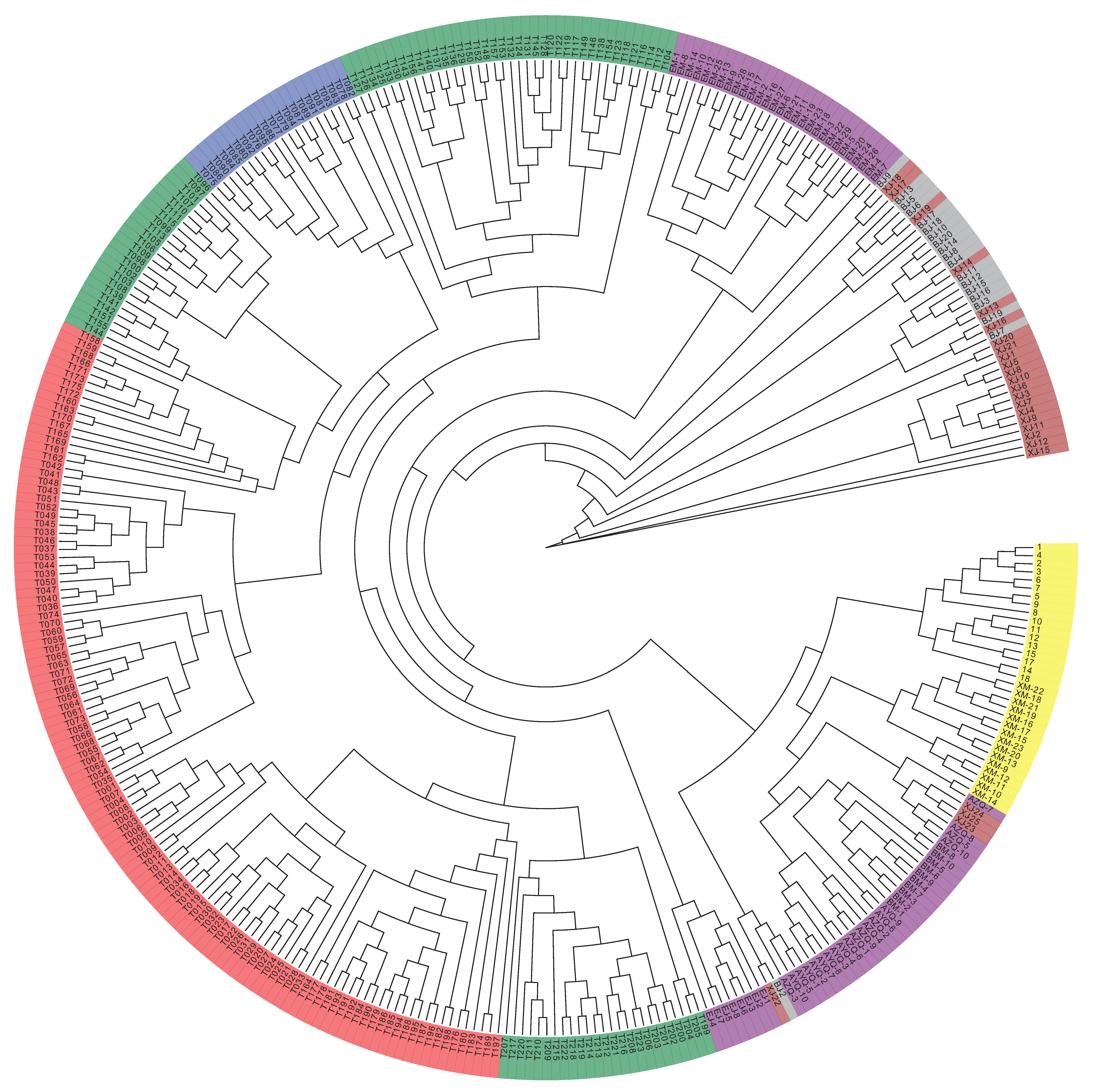
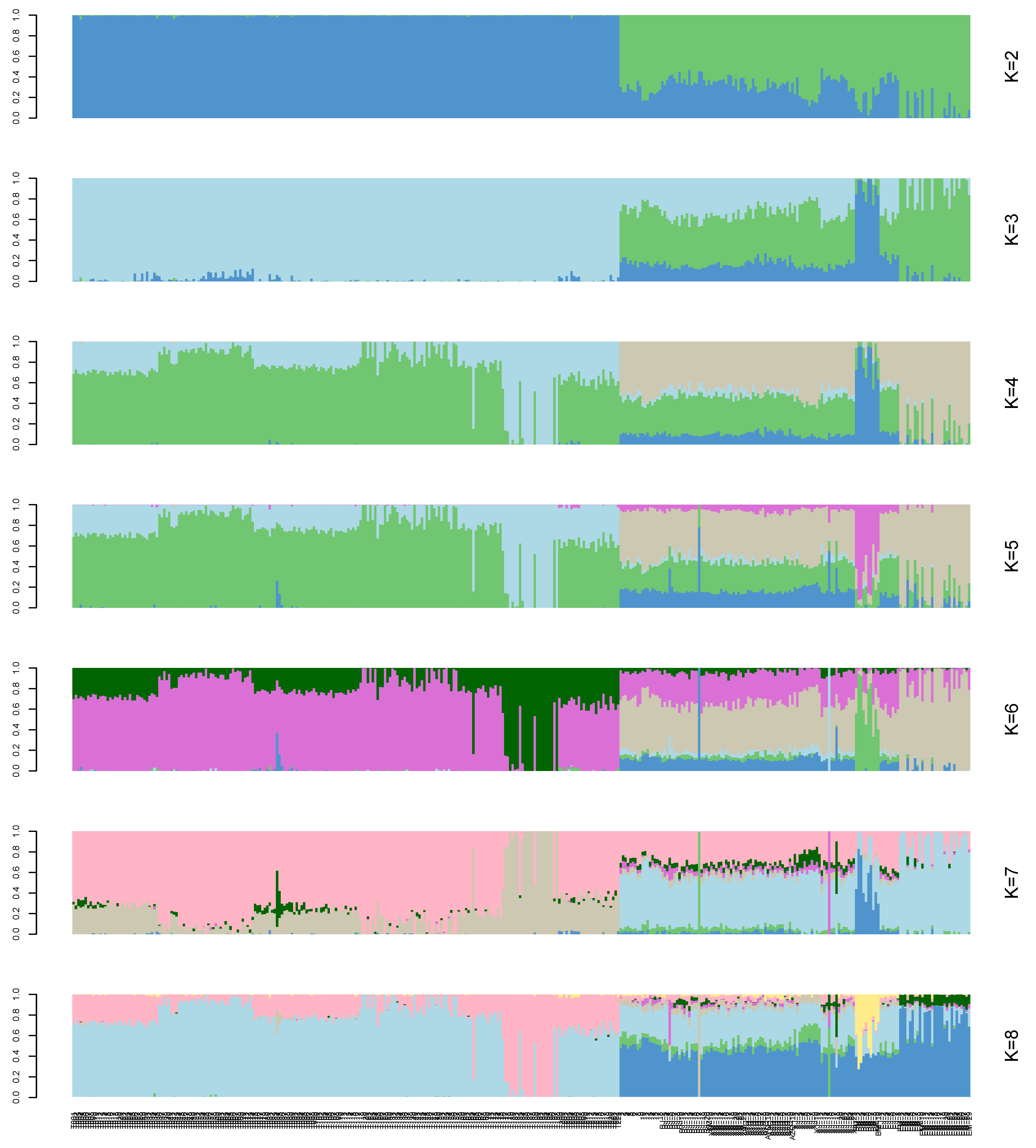
3.4. Analysis of the Population Structure
3.5. GWAS of Phenotypic Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kamatani, Y.; Matsuda, K.; Okada, Y.; Kubo, M.; Hosono, N.; Daigo, Y.; Nakamura, Y.; Kamatani, N. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat. Genet. 2010, 42, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, W.N.; Khalil, J.K.; Al-Shalhat, A.; Al-Mohammad, H. Chemical Composition and Nutritional Quality of Camel Milk. J. Food Sci. 2010, 49, 744–747. [Google Scholar] [CrossRef]
- Ali, A.; Baby, B.; Vijayan, R. From Desert to Medicine: A Review of Camel Genomics and Therapeutic Products. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Almathen, F.; Charruau, P.; Mohandesan, E.; Mwacharo, J.M.; Orozco-terWengel, P.; Pitt, D.; Abdussamad, A.M.; Uerpmann, M.; Uerpmann, H.P.; De Cupere, B.; et al. Ancient and modern DNA reveal dynamics of domestication and cross-continental dispersal of the dromedary. Proc. Natl. Acad. Sci. USA 2016, 113, 6707–6712. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.; Driesch, A.V.D. The two-humped camel (Camelus bactrianus): New light on its distribution, management and medical treatment in the past. J. Zool. 1997, 242, 651–679. [Google Scholar] [CrossRef]
- Ji, R.; Cui, P.; Ding, F.; Geng, J.; Gao, H.; Zhang, H.; Yu, J.; Hu, S.; Meng, H. Monophyletic origin of domestic bactrian camel (Camelus bactrianus) and its evolutionary relationship with the extant wild camel (Camelus bactrianus ferus). Anim. Genet. 2010, 40, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Tulgat, R.; Schaller, G.B. Status and distribution of wild Bactrian camels Camelus bactrianus ferus. Biol. Conserv. 1992, 62, 11–19. [Google Scholar] [CrossRef]
- Sequencing, T.B.; Wang, Z.; Ding, G.; Chen, G.; Sun, Y.; Sun, Z.; Zhang, H.; Wang, L.; Hasi, S.; Zhang, Y.; et al. Genome sequences of wild and domestic bactrian camels. Nat. Commun. 2012, 3, 1202. [Google Scholar]
- Wu, H.; Guang, X.; Al-Fageeh, M.B.; Cao, J.; Pan, S.; Zhou, H.; Zhang, L.; Abutarboush, M.H.; Xing, Y.; Xie, Z.; et al. Camelid genomes reveal evolution and adaptation to desert environments. Nat. Commun. 2014, 5, 5188. [Google Scholar] [CrossRef] [Green Version]
- White, M.E.; Hayward, J.J.; Stokol, T.; Boyko, A.R. Genetic Mapping of Novel Loci Affecting Canine Blood Phenotypes. PLoS ONE 2015, 10, e0145199. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Cui, L.; Huang, T.; Zhang, Z.; Yang, B.; Chen, C.; Huang, L.; Duan, Y. Genome-wide identification of quantitative trait transcripts for blood traits in the liver samples of a White Duroc × Erhualian F2 pig resource population. Physiol. Genom. 2016, 48, 573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Huang, X.; Zeng, Z.; Zhang, W.; Liu, C.; Fang, S.; Huang, L.; Chen, C. Genome-Wide Association Analysis for Blood Lipid Traits Measured in Three Pig Populations Reveals a Substantial Level of Genetic Heterogeneity. PLoS ONE 2015, 10, e0131667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, E.J.; Park, H.B.; Lee, J.B.; Yoo, C.K.; Kim, B.M.; Kim, H.I.; Cho, I.C.; Lim, H.T. Genome-wide association study identifies quantitative trait loci affecting hematological traits in an F2 intercross between Landrace and Korean native pigs. Anim. Genet. 2014, 45, 534–541. [Google Scholar] [CrossRef]
- Nürnberg, S.T.; Rendon, A.; Smethurst, P.A.; Paul, D.S.; Voss, K.; Thon, J.N.; Lloyd-Jones, H.; Sambrook, J.G.; Tijssen, M.R.; Italiano, J.E.; et al. A GWAS sequence variant for platelet volume marks an alternative DNM3 promoter in megakaryocytes near a MEIS1 binding site. Blood 2012, 120, 4859. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhao, Y.; Li, C.; Wang, A.; Zhao, Q.; Li, W.; Guo, Y.; Deng, L.; Zhu, C.; Fan, D.; et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2012, 44, 32. [Google Scholar] [CrossRef]
- Kang, Y.; Sakiroglu, M.; Krom, N.; Stanton-Geddes, J.; Wang, M.; Lee, Y.C.; Young, N.D.; Udvardi, M. Genome-wide association of drought-related and biomass traits with HapMap SNPs in Medicago truncatula. Plant Cell Environ. 2015, 38, 1997–2011. [Google Scholar] [CrossRef]
- Ott, A.; Liu, S.; Schnable, J.C.; Yeh, C.; Wang, K.S.; Schnable, P.S. tGBS® genotyping-by-sequencing enables reliable genotyping of heterozygous loci. Nucleic Acids Res. 2017, 45, e178. [Google Scholar] [CrossRef] [Green Version]
- Underwood, C.J.; Choi, K.; Lambing, C.; Zhao, X.; Serra, H.; Borges, F.; Simorowski, J.; Ernst, E.; Jacob, Y.; Henderson, I.R.; et al. Epigenetic activation of meiotic recombination near Arabidopsis thaliana centromeres via loss of H3K9me2 and non-CG DNA methylation. Genome Res. 2018, 28, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Parker, C.C.; Gopalakrishnan, S.; Carbonetto, P.; Gonzales, N.M.; Leung, E.; Park, Y.J.; Aryee, E.; Davis, J.; Blizard, D.A.; Ackert-Bicknell, C.L.; et al. Genome-wide association study of behavioral, physiological and gene expression traits in outbred CFW mice. Nat. Genet. 2016, 48, 919. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2012, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J. The Sequence Alignment-Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Kai, W.; Mingyao, L.; Hakon, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar]
- An, J.; Won, S.; Lutz, S.M.; Hecker, J.; Lange, C. Effect of population stratification on SNP-by-environment interaction. Genet. Epidemiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Salm, M.P.; Horswell, S.D.; Hutchison, C.E.; Speedy, H.E.; Yang, X.; Liang, L.; Schadt, E.E.; Cookson, W.O.; Wierzbicki, A.S.; Naoumova, R.P.; et al. The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Res. 2012, 22, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Lee, C. A mixed model reduces spurious genetic associations produced by population stratification in genome-wide association studies. Genomics 2015, 105, 191–196. [Google Scholar] [CrossRef]
- Engelman, C.D.; Baurley, J.W.; Chiu, Y.F.; Joubert, B.R.; Lewinger, J.P.; Maenner, M.J.; Murcray, C.E.; Shi, G.; Gauderman, W.J. Detecting Gene-Environment Interactions in Genome-Wide Association Data. Genet. Epidemiol. 2010, 33, S68–S73. [Google Scholar] [CrossRef] [Green Version]
- Park, B.; Koo, S.M.; An, J.; Lee, M.; Kang, H.Y.; Qiao, D.; Cho, M.H.; Sung, J.; Silverman, E.K.; Yang, H.J.; et al. Genome-wide assessment of gene-by-smoking interactions in COPD. Sci. Rep. UK 2018, 8, 9319. [Google Scholar] [CrossRef]
- Kaczensky, P.; Adiya, Y.; von Wehrden, H.; Mijiddorj, B.; Walzer, C.; Güthlin, D.; Enkhbileg, D.; Reading, R.P. Space and habitat use by wild Bactrian camels in the Transaltai Gobi of southern Mongolia. Biol. Conserv. 2014, 169, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Taylor, W.; Shnaider, S.; Abdykanova, A.; Fages, A.; Welker, F.; Irmer, F.; Seguin-Orlando, A.; Khan, N.; Douka, K.; Kolobova, K.; et al. Early pastoral economies along the Ancient Silk Road: Biomolecular evidence from the Alay Valley, Kyrgyzstan. PLoS ONE 2018, 13, e0205646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, K.; Yamamoto, E.; Aya, K. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927. [Google Scholar] [CrossRef] [PubMed]
- De Tayrac, M.; Roth, M.P.; Jouanolle, A.M.; Coppin, H.; Le Gac, G.; Piperno, A.; Férec, C.; Pelucchi, S.; Scotet, V.; Bardou-Jacquet, E.; et al. Genome-wide association study identifies TF as a significant modifier gene of iron metabolism in HFE hemochromatosis. J. Hepatol. 2015, 62, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Jia, C.; Fu, D.; Chu, M.; Ding, X.; Wu, X.; Guo, X.; Pei, J.; Bao, P.; Liang, C.; et al. Analysis of Hematological Traits in Polled Yak by Genome-Wide Association Studies Using Individual SNPs and Haplotypes. Genes 2019, 10, 463. [Google Scholar] [CrossRef] [Green Version]
- Kounis, N.G.; Soufras, G.D.; Tsigkas, G.; Hahalis, G. White blood cell counts, leukocyte ratios, and eosinophils as inflammatory markers in patients with coronary artery disease. Clin. Appl. thromb-Hemost. 2015, 21, 139–143. [Google Scholar] [CrossRef]
- Ellmeier, W.; Taniuchi, I. The role of btb-zinc finger transcription factors during t cell development and in the regulation of t cell-mediated immunity. Curr. Top. Microbiol. 2014, 381, 21–49. [Google Scholar]
- Cartron, P.F.; Petit, E.; Bellot, G.; Oliver, L.; Vallette, F.M. Metaxins 1 and 2, two proteins of the mitochondrial protein sorting and assembly machinery, are essential for bak activation during tnf alpha triggered apoptosis. Cell Signal. 2014, 26, 1928–1934. [Google Scholar] [CrossRef]
- Son, Y.O.; Park, S.; Kwak, J.S.; Won, Y.; Choi, W.S.; Rhee, J.; Chun, C.H.; Ryu, J.H.; Kim, D.K.; Choi, H.S.; et al. Estrogen-related receptor γ causes osteoarthritis by upregulating extracellular matrix-degrading enzymes. Nat. Commun. 2017, 8, 2133. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean | Standard Deviation | Minimum | Maximum |
|---|---|---|---|---|
| White blood cell count, 109 cells/L (WBC) | 21.95 | 11.07 | 4.01 | 65.5 |
| Red blood cell count, 1012 cells/L (RBC) | 8.64 | 2.11 | 4.8 | 28 |
| Hemoglobin, g/L (HGB) | 128.91 | 21.42 | 66 | 190 |
| Mean corpuscular volume, fL (MCV) | 41.83 | 4.67 | 34.5 | 58 |
| Hematocrit, % (HCT) | 0.35 | 0.13 | 0.16 | 1.4 |
| Mean corpuscular hemoglobin, pg (MCH) | 16.47 | 3.93 | 5.4 | 28.5 |
| Mean corpuscular hemoglobin concentration, g/L (MCHC) | 395 | 84.85 | 98 | 620 |
| Red cell distribution width, % (RDW_SD) | 56.72 | 7.1 | 45.1 | 91.3 |
| Red cell distribution width, % (RDW_CV) | 57.72 | 6.12 | 31.2 | 49.9 |
| Platelet count, 109 cells/L (PLT) | 372.58 | 183.26 | 87.5 | 1007.5 |
| Mean platelet volume, fL (MPV) | 15.72 | 2.78 | 6.1 | 19.9 |
| Plateletcrit, % (PCT) | 0.58 | 0.33 | 0.11 | 1.98 |
| Platelet distribution width, % (PDW) | 0.16 | 0.06 | 0.11 | 0.25 |
| Aspartate aminotransferase, U/L (AST) | 105.5 | 25.15 | 58.8 | 185.8 |
| Alanine aminotransferase, U/L (ALT) | 15.43 | 4.13 | 25.4 | 4.2 |
| Glutamyl transpeptidase, U/L (GGT) | 25.53 | 12.69 | 6.5 | 83.2 |
| Total cholesterol, mmol/L (CHO) | 1.49 | 0.5 | 0.78 | 5.29 |
| Items | Category | Number of SNPs |
|---|---|---|
| Upstream | 2671 | |
| Exonic | Stop gain | 61 |
| Stop loss | 5 | |
| Synonymous | 1380 | |
| Non-synonymous | 1496 | |
| Intronic | 155,104 | |
| Splicing | 37 | |
| Downstream | 3099 | |
| upstream/downstream | 28 | |
| Intergenic | 384,757 | |
| ts | 332,710 | |
| tv | 216,035 | |
| ts/tv | 1.540 | |
| Total | 548,745 |
| Trait | Scaffold | Peak Position | Reference | alt | −log p-Value | Candidate Gene | Annotation |
|---|---|---|---|---|---|---|---|
| WBC | gi|558497537|ref|NW_006212822.1| | 30,297 | T | C | 7.407936107 | ZNF772 | Zinc finger protein 772 |
| RBC | gi|558499008|ref|NW_006211351.1| | 31,481 | C | T | 12.64681147 | MTX2 | Metaxin 2 |
| HGB | gi|558499111|ref|NW_006211248.1| | 8,581,139 | C | G | 7.761062626 | ESRRG | Estrogen-related receptor gamma |
| MCV | gi|558499592|ref|NW_006210767.1| | 2,375,652 | A | G | 7.192791086 | MEI4 | Meiotic double-stranded break formation protein 4 |
| HCT | gi|558499008|ref|NW_006211351.1| | 31,481 | C | T | 11.84901895 | IL11 | Interleukin 11 |
| MCH | gi|558498226|ref|NW_006212133.1| | 120,804 | T | G | 6.462211621 | FRMPD4 | FERM and PDZ domain containing 4 |
| RDW_SD | gi|558499308|ref|NW_006211051.1| | 1,267,292 | A | G | 11.82658732 | GABPA | GA binding protein transcription factor alpha subunit 60 kDa |
| RDW_CV | gi|558499308|ref|NW_006211051.1| | 1,267,292 | A | G | 9.337752481 | GABPA | GA binding protein transcription factor alpha subunit 60 kDa |
| MPV | gi|558500051|ref|NW_006210308.1| | 893,732 | T | A | 9.240644582 | NTF4 | Neurotrophin-4 |
| PCT | gi|558498489|ref|NW_006211870.1| | 319,343 | G | A | 6.093058393 | CRYBG3 | Beta-gamma crystallin domain containing 3 |
| PDW | gi|558498782|ref|NW_006211577.1| | 5,532,111 | T | G | 14.12056347 | ENPP5 | Ectonucleotide pyrophosphatase/phosphodiesterase 5 (putative) |
| GGT | gi|558499927|ref|NW_006210432.1| | 871,319 | C | A | 8.005790752 | COL16A1 | Collagen type XVI alpha 1 |
| CHO | gi|558498607|ref|NW_006211752.1| | 431,610 | G | A | 15.38590792 | CD207 | ATPase H+ transporting lysosomal 56/58kDa V1 subunit B1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, F.; Ming, L.; Si, R.; Yi, L.; He, J.; Ji, R. A Genome-Wide Association Study Identifies Quantitative Trait Loci Affecting Hematological Traits in Camelus bactrianus. Animals 2020, 10, 96. https://doi.org/10.3390/ani10010096
Guo F, Ming L, Si R, Yi L, He J, Ji R. A Genome-Wide Association Study Identifies Quantitative Trait Loci Affecting Hematological Traits in Camelus bactrianus. Animals. 2020; 10(1):96. https://doi.org/10.3390/ani10010096
Chicago/Turabian StyleGuo, Fucheng, Liang Ming, Rendalai Si, Li Yi, Jing He, and Rimutu Ji. 2020. "A Genome-Wide Association Study Identifies Quantitative Trait Loci Affecting Hematological Traits in Camelus bactrianus" Animals 10, no. 1: 96. https://doi.org/10.3390/ani10010096
APA StyleGuo, F., Ming, L., Si, R., Yi, L., He, J., & Ji, R. (2020). A Genome-Wide Association Study Identifies Quantitative Trait Loci Affecting Hematological Traits in Camelus bactrianus. Animals, 10(1), 96. https://doi.org/10.3390/ani10010096