Estimation of Variance Components and Genomic Prediction for Individual Birth Weight Using Three Different Genome-Wide SNP Platforms in Yorkshire Pigs

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Descriptive Statistics of the Phenotype and Pedigree
2.2. Genotypic Data Editing and Imputation
2.3. Estimating the Variance Components and the Genetic Parameters
2.3.1. Model Definition
2.3.2. The Likelihood Ratio Test
2.4. Deregressed Estimated Breeding Values of the Response Variables in Genomic Analysis
2.5. Statistical Method for Estimating SNP Effects
2.6. Genomic Prediction Accuracy under Five-Fold Cross-Validation
3. Results and Discussion
3.1. Heritability and the Optimized Model
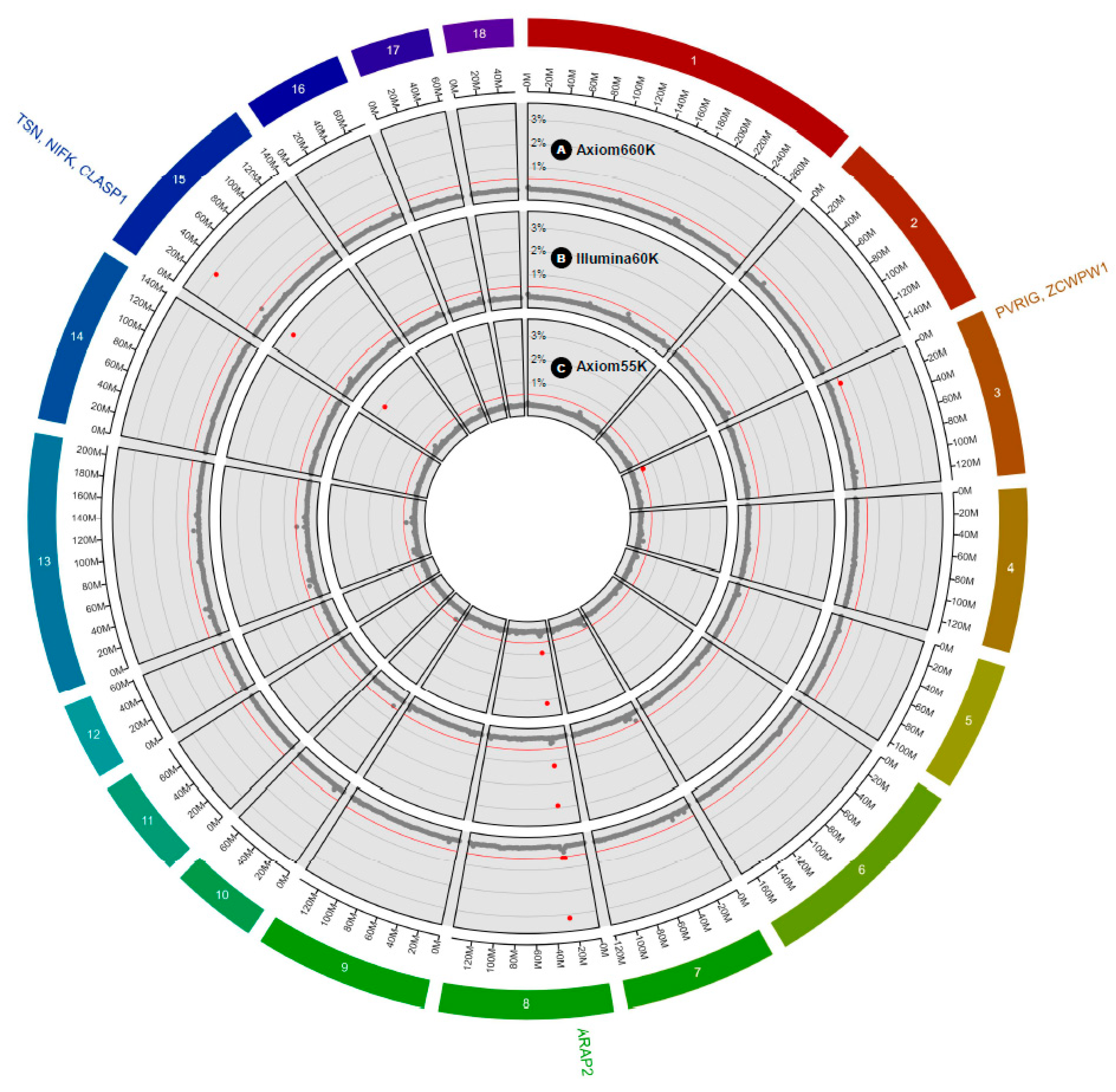
3.2. GWAS of the Individual Birth Weight Trait
3.3. Accuracy of the Direct Genomic Values
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bakoev, S.; Getmantseva, L.; Bakoev, F.; Kolosova, M.; Gabova, V.; Kolosov, A.; Kostyunina, O. Survey of SNPs Associated with Total Number Born and Total Number Born Alive in Pig. Genes 2020, 11, 491. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Konta, A.; Kimata, M.; Ishii, K.; Uemoto, Y.; Satoh, M. Estimation of genetic parameters for farrowing traits in purebred Landrace and Large White pigs. Anim. Sci. J. 2019, 90, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Gomez, M.; Garcia-Contreras, C.; Pesantez-Pacheco, J.L.; Torres-Rovira, L.; Heras-Molina, A.; Astiz, S.; Ovilo, C.; Isabel, B.; Gonzalez-Bulnes, A. Differential Effects of Litter Size and Within-Litter Birthweight on Postnatal Traits of Fatty Pigs. Animals 2020, 10, 870. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, O.; Baxter, E.M.; Lawlor, P.G.; Boyle, L.A.; O’Driscoll, K. A single dose of fat-based energy supplement to light birth weight pigs shortly after birth does not increase their survival and growth. Animals 2019, 9, 227. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, B.; Yang, M.; Han, H.; Chen, T.; Wei, Q.; Miao, Z.; Yin, L.; Wang, R.; Shen, J.; et al. Genome-Wide Association Study and Fine Mapping Reveals Candidate Genes for Birth Weight of Yorkshire and Landrace Pigs. Front. Genet. 2020, 11, 183. [Google Scholar] [CrossRef]
- Feldpausch, J.A.; Jourquin, J.; Bergstrom, J.R.; Bargen, J.L.; Bokenkroger, C.D.; Davis, D.L.; Gonzalez, J.M.; Nelssen, J.L.; Puls, C.L.; Trout, W.E.; et al. Birth weight threshold for identifying piglets at risk for preweaning mortality. Transl. Anim. Sci. 2019, 3, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Quiniou, N.; Dagorn, J.; Gaudre, D. Variation of piglets birth weight and consequences on subsequent performance. Livest. Prod. Sci. 2002, 78, 63–70. [Google Scholar] [CrossRef]
- Wolf, J.; Zakova, E.; Groeneveld, E. Within-litter variation of birth weight in hyperprolific Czech Large White sows and its relation to litter size traits, stillborn piglets and losses until weaning. Livest. Sci. 2008, 115, 195–205. [Google Scholar] [CrossRef]
- Alves, K.; Schenkel, F.S.; Brito, L.F.; Robinson, A. Estimation of direct and maternal genetic parameters for individual birth weight, weaning weight, and probe weight in Yorkshire and Landrace pigs. J. Anim. Sci. 2018, 96, 2567–2578. [Google Scholar] [CrossRef]
- Engblom, L.; Calderon Diaz, J.A.; Nikkila, M.; Gray, K.; Harms, P.; Fix, J.; Tsuruta, S.; Mabry, J.; Stalder, K. Genetic analysis of sow longevity and sow lifetime reproductive traits using censored data. J. Anim. Breed Genet. 2016, 133, 138–144. [Google Scholar] [CrossRef]
- Kaufmann, D.; Hofer, A.; Bidanel, J.P.; Kunzi, N. Genetic parameters for individual birth and weaning weight and for litter size of Large White pigs. J. Anim. Breed. Genet. 2000, 117, 121–128. [Google Scholar] [CrossRef]
- Roehe, R. Genetic determination of individual birth weight and its association with sow productivity traits using Bayesian analyses. J. Anim. Sci. 1999, 77, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Te Pas, M.F.; Soumillion, A.; Harders, F.L.; Verburg, F.J.; van den Bosch, T.J.; Galesloot, P.; Meuwissen, T.H. Influences of myogenin genotypes on birth weight, growth rate, carcass weight, backfat thickness, and lean weight of pigs. J. Anim. Sci. 1999, 77, 2352–2356. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Li, N.; Fan, X.Z.; Xiao, L.R.; Xiang, R.L.; Hu, X.X.; Du, L.X.; Wu, C.X. Associations of T→A mutation in the promoter region of myostatin gene with birth weight in Yorkshire pigs. Asian-Australas. J. Anim. Sci. 2002, 15, 1543–1545. [Google Scholar] [CrossRef]
- Tomás, A.; Casellas, J.; Ramírez, O.; Pérez-Enciso, M.; Rodriguez, C.; Noguera, J.; Sánchez, A. Polymorphisms of the porcine dopamineβ-hydroxylase gene and their relation to reproduction and piglet survivability in an Iberian× Meishan F2 intercross. Anim. Genet. 2006, 37, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.J.; Goddard, M.E. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolc, A.; Arango, J.; Settar, P.; Fulton, J.E.; O’Sullivan, N.P.; Preisinger, R.; Habier, D.; Fernando, R.; Garrick, D.J.; Hill, W.G.; et al. Genome-wide association analysis and genetic architecture of egg weight and egg uniformity in layer chickens. Anim. Genet. 2012, 43 (Suppl. 1), 87–96. [Google Scholar] [CrossRef]
- Lourenco, D.A.L.; Tsuruta, S.; Fragomeni, B.O.; Masuda, Y.; Aguilar, I.; Legarra, A.; Bertrand, J.K.; Amen, T.S.; Wang, L.; Moser, D.W.; et al. Genetic evaluation using single-step genomic best linear unbiased predictor in American Angus. J. Anim. Sci. 2015, 93, 2653–2662. [Google Scholar] [CrossRef] [Green Version]
- Riggio, V.; Portolano, B. Genetic selection for reduced somatic cell counts in sheep milk: A review. Small Rumin. Res. 2015, 126, 33–42. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Park, J.E.; Moon, S.H.; Choi, S.W.; Go, G.W.; Lim, D.; Kim, J.M. Genome-wide association study and genomic predictions for exterior traits in Yorkshire pigs. J. Anim. Sci. 2019, 97, 2793–2802. [Google Scholar] [CrossRef]
- Zhang, C.; Patterson, J.; Tsoi, S.; Dyck, M.; Plastow, G.; Foxcroft, G. GWAS in production nucleus sows using a 650K SNP Chip to explore component traits underlying a repeatable low litter birth weight phenotype. In Proceedings of the World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018; p. 567. [Google Scholar]
- Wang, Y.; Ding, X.; Tan, Z.; Xing, K.; Yang, T.; Pan, Y.; Wang, Y.; Mi, S.; Sun, D.; Wang, C. Genome-wide association study for reproductive traits in a Large White pig population. Anim. Genet. 2018, 49, 127–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.F.; Zhou, X.; Michal, J.J.; Ding, B.; Li, R.; Jiang, Z.H. Genome Wide Screening of Candidate Genes for Improving Piglet Birth Weight Using High and Low Estimated Breeding Value Populations. Int. J. Biol. Sci. 2014, 10, 236–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargolzaei, M.; Chesnais, J.P.; Schenkel, F.S. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 2014, 15, 478. [Google Scholar] [CrossRef] [Green Version]
- Gilmour, A.; Gogel, B.; Cullis, B.; Welham, S.; Thompson, R. ASReml User Guide Release 4.1 Structural Specification; VSN International Ltd.: Hemel Hempstead, UK, 2015. [Google Scholar]
- Bouwman, A.; Bergsma, R.; Duijvesteijn, N.; Bijma, P. Maternal and social genetic effects on average daily gain of piglets from birth until weaning. J. Anim. Sci. 2010, 88, 2883–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Kizilkaya, K.; Fernando, R.; Garrick, D. Genomic prediction of simulated multibreed and purebred performance using observed fifty thousand single nucleotide polymorphism genotypes. J. Anim. Sci. 2010, 88, 544–551. [Google Scholar] [CrossRef] [Green Version]
- Garrick, D.J.; Fernando, R.L. Implementing a QTL detection study (GWAS) using genomic prediction methodology. In Genome-Wide Association Studies and Genomic Prediction; Springer: Berlin/Heidelberg, Germany, 2013; pp. 275–298. [Google Scholar]
- R Development Core. A Language ans Environment for Statistical Computing; R Found Stat Comput: Vienna, Austria, 2018; Volume 2. [Google Scholar]
- Kruuk, L.E.; Hadfield, J.D. How to separate genetic and environmental causes of similarity between relatives. J. Evol. Biol. 2007, 20, 1890–1903. [Google Scholar] [CrossRef]
- Yoon, H.-Y.; Miura, K.; Cuthbert, E.J.; Davis, K.K.; Ahvazi, B.; Casanova, J.E.; Randazzo, P.A. ARAP2 effects on the actin cytoskeleton are dependent on Arf6-specific GTPase-activating-protein activity and binding to RhoA-GTP. J. Cell Sci. 2006, 119, 4650–4666. [Google Scholar] [CrossRef] [Green Version]
- Bunnell, T.M.; Burbach, B.J.; Shimizu, Y.; Ervasti, J.M. β-Actin specifically controls cell growth, migration, and the G-actin pool. Mol. Biol. Cell 2011, 22, 4047–4058. [Google Scholar] [CrossRef]
- Ikeuchi, Y.; Imanishi, A.; Sudo, K.; Fukunaga, T.; Yokoi, A.; Matsubara, L.; Goto, C.; Fukuoka, T.; Kuronuma, K.; Kono, R. Translin modulates mesenchymal cell proliferation and differentiation in mice. Biochem. Biophys. Res. Commun. 2018, 504, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, T.; Goddard, M. Accurate prediction of genetic values for complex traits by whole-genome resequencing. Genetics 2010, 185, 623–631. [Google Scholar] [CrossRef]
- Brito, F.V.; Neto, J.B.; Sargolzaei, M.; Cobuci, J.A.; Schenkel, F.S. Accuracy of genomic selection in simulated populations mimicking the extent of linkage disequilibrium in beef cattle. BMC Genet. 2011, 12, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbe, M.; Hayes, B.J.; Matukumalli, L.K.; Goswami, S.; Bowman, P.J.; Reich, C.M.; Mason, B.A.; Goddard, M.E. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy Sci. 2012, 95, 4114–4129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kachman, S.D.; Spangler, M.L. The impact of training strategies on the accuracy of genomic predictors in United States Red Angus cattle. J. Anim Sci. 2017, 95, 3406–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, Y.; Cho, E.; Cho, K.; Sa, S.; Kim, Y.; Choi, J.; Kim, J.; Hong, J.; Choi, T. Genomic Analysis Using Bayesian Methods under Different Genotyping Platforms in Korean Duroc Pigs. Animals 2020, 10, 752. [Google Scholar] [CrossRef]
- Gunia, M.; Saintilan, R.; Venot, E.; Hoze, C.; Fouilloux, M.N.; Phocas, F. Genomic prediction in French Charolais beef cattle using high-density single nucleotide polymorphism markers. J. Anim. Sci. 2014, 92, 3258–3269. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Kemp, R.A.; Stothard, P.; Wang, Z.; Boddicker, N.; Krivushin, K.; Dekkers, J.; Plastow, G. Genomic evaluation of feed efficiency component traits in Duroc pigs using 80K, 650K and whole-genome sequence variants. Genet. Sel. Evol. 2018, 50, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Perez, P.; de los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.M.; Garrick, D. Increasing the accuracy of genomic prediction in pure-bred Limousin beef cattle by including cross-bred Limousin data and accounting for an F94L variant in MSTN. Anim. Genet. 2019, 50, 621–633. [Google Scholar]

{kind=link}
| Clusters | No. of Animals | inBreC 1 | amax_within2 | amax_between3 | aij_within4 | aij_between5 |
|---|---|---|---|---|---|---|
| 1 | 99 | 0.028 | 0.524 | 0.393 | 0.243 | 0.061 |
| 2 | 133 | 0.015 | 0.497 | 0.344 | 0.177 | 0.047 |
| 3 | 192 | 0.008 | 0.446 | 0.348 | 0.043 | 0.032 |
| 4 | 145 | 0.014 | 0.441 | 0.409 | 0.089 | 0.050 |
| 5 | 109 | 0.001 | 0.476 | 0.402 | 0.132 | 0.031 |
| Avg. | 0.013 | 0.477 | 0.379 | 0.137 | 0.044 |
| Model 1 | Variance Components 2 | h2 ± SE | LogL 3 | LRT 4 | Model Comparison5 | P 6 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Equation (1) | 0.036 | - | - | 0.036 | 0.072 | 0.50 ± 0.013 | 2156.38 | 3678.84 | 1 vs. 4 | <0.0001 |
| Equation (2) | 0.015 | 0.018 | - | 0.039 | 0.071 | 0.21 ± 0.018 | 3140.04 | 2695.18 | 2 vs. 4 | <0.0001 |
| Equation (3) | 0.022 | - | 0.020 | 0.027 | 0.069 | 0.32 ± 0.017 | 5581.12 | 254.1 | 3 vs. 4 | <0.0001 |
| Equation (4) | 0.009 | 0.010 | 0.017 | 0.030 | 0.065 | 0.13 ± 0.019 | 5835.22 | |||
| Genotyping Platform | SSC_Mb | GV (%) 1 | Informative SNP | Position (Mb) | Genetic Effect | Model Frequency | Region Annotation | Gene Annotation 2 |
|---|---|---|---|---|---|---|---|---|
| Axiom Porcine 55K | 8_27 | 3.17 | AX-116342380 | 27.89 | −0.004 | 0.104 | Intronic | ARAP2 |
| AX-116690854 | 27.84 | 0.002 | 0.056 | Intronic | ARAP2 | |||
| AX-116342258 | 27.44 | −0.002 | 0.050 | Intergenic | ARAP2(dist = 333210) | |||
| AX-116342267 | 27.48 | −0.002 | 0.050 | Intergenic | ARAP2(dist = 333210) | |||
| AX-116342268 | 27.48 | −0.001 | 0.049 | Intergenic | ARAP2(dist = 333210) | |||
| 15_29 | 2.95 | AX-116536263 | 29.71 | 0.005 | 0.118 | Intronic | TSN | |
| AX-116674286 | 29.64 | 0.004 | 0.093 | Intergenic | TSN(dist = 37022) | |||
| AX-116536281 | 29.78 | −0.003 | 0.074 | Intronic | NIFK | |||
| 8_26 | 0.96 | AX-116690835 | 26.54 | 0.002 | 0.067 | Intergenic | ARAP2(dist = 1271371) | |
| 3_8 | 0.57 | AX-116718059 | 8.28 | 0.003 | 0.061 | Intergenic | PVRIG(dist = 49034), ZCWPW1(dist = 41321) | |
| Illumina 60K | 15_29 | 3.31 | DBWU0000855 | 29.71 | 0.006 | 0.1299 | Intronic | TSN |
| H3GA0044096 | 29.64 | 0.004 | 0.0934 | Intergenic | TSN(dist = 37022) | |||
| ALGA0084705 | 29.73 | −0.002 | 0.0649 | Intronic | NIFK | |||
| ALGA0084700 | 29.82 | -0.001 | 0.0525 | Intronic | CLASP1 | |||
| 8_27 | 2.91 | ALGA0047127 | 27.89 | −0.005 | 0.1284 | Intronic | ARAP2 | |
| ALGA0047120 | 27.86 | 0.002 | 0.0726 | Intronic | ARAP2 | |||
| ALGA0047098 | 27.48 | −0.002 | 0.0636 | Intergenic | ARAP2(dist = 294518) | |||
| ALGA0047102 | 27.52 | 0.001 | 0.0557 | Intergenic | ARAP2(dist = 256059) | |||
| 8_26 | 1.24 | INRA0029430 | 26.50 | 0.002 | 0.0783 | Intergenic | ARAP2(dist = 1271371) | |
| 7_9 | 0.41 | ALGA0107233 | 9.29 | 0.001 | 0.0493 | Intronic | PHACTR1 | |
| Axiom porcine 660K | 8_27 | 3.16 | AX-116342374 | 27.87 | 0.001 | 0.021 | Intronic | ARAP2 |
| AX-116342286 | 27.54 | −0.001 | 0.016 | Intergenic | ARAP2(dist = 229771) | |||
| AX-116342230 | 27.31 | 0.001 | 0.012 | Intergenic | ARAP2(dist = 456854) | |||
| AX-116342377 | 27.88 | 0.001 | 0.012 | Intronic | ARAP2 | |||
| 15_29 | 2.89 | AX-116762423 | 29.65 | −0.001 | 0.023 | Intergenic | TSN(dist = 22517) | |
| AX-116744904 | 29.65 | 0.001 | 0.022 | Intergenic | TSN(dist=22517) | |||
| AX-116536258 | 29.71 | −0.001 | 0.014 | Intronic | NIFK | |||
| AX-116536264 | 29.72 | −0.001 | 0.012 | Intronic | NIFK | |||
| AX-116536266 | 29.73 | 0.001 | 0.012 | Intronic | NIFK |
| Trait 1 | Response Variables 2 | Genotyping Platforms | π 3 | BayesB | BayesC |
|---|---|---|---|---|---|
| IBW | DEBVexcPA | Axiom55K | 0.99 | 0.188 (0.014) | 0.178 (0.013) |
| Illumina60Kv2 | 0.99 | 0.168 (0.012) | 0.157 (0.012) | ||
| Axiom660K | 0.999 | 0.163 (0.013) | 0.150 (0.013) | ||
| DEBVincPA | Axiom55K | 0.99 | 0.261 (0.012) | 0.252 (0.013) | |
| Illumina60Kv2 | 0.99 | 0.224 (0.013) | 0.210 (0.013) | ||
| Axiom660K | 0.999 | 0.223 (0.012) | 0.215 (0.013) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Lee, S.-M.; Lim, B.; Park, J.; Song, K.-L.; Jeon, J.-H.; Na, C.-S.; Kim, J.-M. Estimation of Variance Components and Genomic Prediction for Individual Birth Weight Using Three Different Genome-Wide SNP Platforms in Yorkshire Pigs. Animals 2020, 10, 2219. https://doi.org/10.3390/ani10122219
Lee J, Lee S-M, Lim B, Park J, Song K-L, Jeon J-H, Na C-S, Kim J-M. Estimation of Variance Components and Genomic Prediction for Individual Birth Weight Using Three Different Genome-Wide SNP Platforms in Yorkshire Pigs. Animals. 2020; 10(12):2219. https://doi.org/10.3390/ani10122219
Chicago/Turabian StyleLee, Jungjae, Sang-Min Lee, Byeonghwi Lim, Jun Park, Kwang-Lim Song, Jung-Hwan Jeon, Chong-Sam Na, and Jun-Mo Kim. 2020. "Estimation of Variance Components and Genomic Prediction for Individual Birth Weight Using Three Different Genome-Wide SNP Platforms in Yorkshire Pigs" Animals 10, no. 12: 2219. https://doi.org/10.3390/ani10122219
APA StyleLee, J., Lee, S.-M., Lim, B., Park, J., Song, K.-L., Jeon, J.-H., Na, C.-S., & Kim, J.-M. (2020). Estimation of Variance Components and Genomic Prediction for Individual Birth Weight Using Three Different Genome-Wide SNP Platforms in Yorkshire Pigs. Animals, 10(12), 2219. https://doi.org/10.3390/ani10122219