1. Introduction
Chlamydia psittaci (
C. psittaci) is an obligate, intracellular, Gram-negative bacterium and an economically relevant pathogen in poultry and pet birds, where it causes psittacosis/ornithosis, as well as being a human pathogen that causes atypical pneumonia after zoonotic transmission [
1]. In the period from 1890 to 1930, human psittacosis outbreaks in Europe and America can all be attributed to contact with sick birds [
2]. Later, due to improved knowledge on etiology and epidemiology, as well as the use of antimicrobials in therapy, large outbreaks became rare exceptions. However, what should still not be ignored is that chlamydiosis is still widespread and represents a major factor of economic loss to the poultry industry, as well as a permanent risk for zoonotic transmission to human [
3]. Apart from overt clinical manifestations, latent
C. psittaci infection can cause recurrent and chronic diseases that have an adverse effect on the production performance of animals. Recently,
C. psittaci prevalence in birds has been reported around the world. For example, Dickx found 58.0% prevalence in feral Canada geese (Branta canadensis) in Belgium [
4]; meanwhile, Cong’s study revealed a high
C. psittaci seroprevalence in pet birds, market-sold adult chickens, ducks, and pigeons in north-western China [
5,
6]. In our previous survey,
C. psittaci-specific serum antibodies and antigen were detected in 10.0% and 26.7% of the studied birds, respectively, suggesting that
C. psittaci prevalence in Beijing is like that reported in European cities. Moreover, the highly positive antigens in pigeon fanciers suggest that exposure and possible zoonotic transmission of
C. psittaci from racing pigeons to humans highlights an ongoing form of transmission [
7]. The study of Wang et al. demonstrated the existence (22.22%) of
C. psittaci infection in pigeons in northern China [
8].
C. psittaci causes respiratory and systemic infection in birds, and continues for a long time under unfavorable conditions.
C. psittaci and other pathogens can cause mixed infection in birds and poultry [
9]. There are numerous reports that viruses and bacteria often act synergistically in causing diseases in humans or animals. In recent years, with the continuous expansion of the poultry industry, the incidence of various diseases has increased year by year, and the mixed infection of
Chlamydia and other pathogens seriously affects the diagnosis and prevention of poultry disease and causes great economic loss to the poultry industry.
The H9 subtype of avian influenza virus (AIV) is one of the subtypes most frequently found in circulation in domestic chickens [
10]. H9N2 influenza virus, a low pathogenic avian influenza virus subtype, has become endemic in different types of domestic poultry in multiple countries due to the occurrence of virulent strains, resulting in great economic losses, such as reduced egg production and high mortality associated with coinfection with other pathogens [
11]. What is worse is that more human infections with H9N2 have been reported since 2014. In a previous study, the results showed that poultry workers had an overall H9N2 seroprevalence of 1.87% and a seroprevalence of 8.78/1000 person-years, which is significantly higher than those of H7N9 and H5N1 [
12]. More importantly, H9N2 has particularly significant implications due to its widespread circulation in domestic poultry, especially in the presence of other coinfecting pathogens. For example, coinfection with
Escherichia coli (
E. coli) and H9N2 caused more serious synergistic pathogenic effects, and indicates the role of both pathogens as complicating factors in poultry infections [
13]. Some researchers have also studied the effect of two major pathogens—namely, H9N2 and avian infectious bronchitis (IBV)—in multiple infections [
14].
Because
C. psittaci and H9N2 are not highly pathogenic, the extent of infection in poultry and humans is likely to remain underestimated. However, these two pathogens are easily mixed with other pathogens, and thus can aggravate diseases. More recently, it was reported that
C. psittaci and H9N2 often cause mixed infections in clinic. Our previous study found that
C. psittaci infection increases the mortality of avian influenza virus H9N2 by suppressing the host immune response [
15]. In addition, coinfection of
C. psittaci with H9N2,
Ornithobacterium rhinotracheale (ORT), and
Aspergillus fumigatus (
A. fumigatus) contributes to severe pneumonia and a high mortality in specific pathogen-free (SPF) chickens, explaining why severe avian airsacculitis is prevalent in the winter season in northern China [
16].
Macrophages play critical roles in innate and adaptive immunity against chlamydial infections. The depletion of macrophages from mice prior to infection with
Chlamydia muridarum (
C. muridarum) and
C. psittaci results in increased morbidity and pathogen burdens [
17,
18]. Macrophage activities may not contribute to pathogen clearance, because
C. psittaci is able to survive and deliver to other tissues by using the macrophage as a “vehicle” [
19]. Macrophages activated by interferon gamma (IFN-γ) and lipopolysaccharide (LPS) or other microbial pathogen-associated molecular patterns (PAMPs) are associated with increased mortality of macrophages, nitric oxide (NO) production, secretions of inducible nitric oxide synthase (iNOS), and pro-inflammatory cytokines [
20]. In this study, we illustrate the pathogenic mechanism of coinfection with
C. psittaci and H9N2 in the chicken macrophage cell line HD11. Based on previous animal studies, we postulate that
C. psittaci infection might be beneficial for the replication of H9N2 in HD11 macrophages by impairing the macrophage functions.
2. Materials and Methods
2.1. Cells and Virus
The chicken macrophage HD11 cells were kindly provided by Prof. Jian Qiao (College of Veterinary Medicine, China Agricultural University, Beijing, China). HD11 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, United States) supplemented with 10% fetal bovine serum (FBS) (Gibco, United States), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, United States) at pH 7.2, and were kept at 37 °C with 5% carbon oxide (CO
2). The Buffalo Green Monkey (BGM) cell-adapted
C. psittaci 6BC strain used in the current study was housed in our laboratory. The
C. psittaci 6BC standard strain was kindly provided by Prof. Yimou Wu (Institute of Pathogenic Biology, University of South China, Hengyang, Hunan Province, China) and the AIV H9N2/chicken/Shandong/2011 was isolated from broilers as described previously [
21]. The susceptibility of HD11 cells to
C. psittaci and H9N2 was measured by morphological changes, growth curves using 50% tissue culture infective doses (TCID
50), and indirect immunofluorescence assay (IFA).
2.2. Virus and Bacteria Titration
The H9N2 AIV (H9N2/chicken/Shandong/2011) chicken embryo allantoic fluid was diluted 1000 times; then, 9~11-day-old SPF chicken embryos were inoculated with 0.2 mL and incubated in 37 °C incubators. After 24 h, the dead chicken embryos were abandoned and the rest were incubated for 48 h in the thermostat. After being at 4 °C refrigerators overnight, the chicken embryo uranic fluid was tested under aseptic conditions, and the hemagglutination-inhibition (HI) titers was measured—it was ≥ 7.0 (log
2) in the uranic solution and saved at −80 °C. Here, TCID
50 was applied to the HD11 cells to quantitate the virus titers, as described previously [
22]. The HD11 cells were cultured in 96-well plates, and 10-fold dilutions of the virus were prepared in DMEM supplemented with 2% FBS. The cultured cells were infected with the virus and then observed daily for cytopathic effects (CPE). The final virus titers were calculated by the Reed–Muench method to be 10
3.86 TCID
50/mL.
As for C. psittaci, the BGM cells were seeded on round coverslips and cultured in growth medium consisting of minimal essential medium (MEM) supplemented with 5% fetal calf serum (FCS). Ten-fold dilutions of the inoculum were centrifuged at 3400× g for 1 h at 37 °C, and then incubated for 2 h at 37 °C. The growth medium was subsequently replaced by serum-free medium. After 48 h of incubation, the cells were fixed in absolute methanol for 10 min. Chlamydial inclusions were detected by direct immunofluorescence using a monoclonal antibody conjugated to fluorescein isothiocyanate (FITC), diluted 1:5 in phosphate-buffered saline (PBS), at pH 7.4 (Imagen Chlamydia, Oxoid, France). The number of inclusion-forming units (IFU) per mL was assessed by counting the total number of inclusions on the whole coverslip of a countable inoculum dilution. A final titer of 4.05 × 108 IFU/mL was determined for the inoculum.
2.3. Experimental Design
The HD11 cells were cultured in DMEM with 10% FBS, seeded in a six-well cell culture plate at a concentration of 1 × 106 per well, and then kept at 37 °C with 5% CO2 for 24 h. The coinfection group cells were infected with C. psittaci and H9N2 simultaneously. The C. psittaci- and H9N2-only infection groups were only infected with C. psittaci or H9N2, respectively. The infection doses of the two pathogens were both at a multiplicity of infection (MOI).
2.4. Quantitative Real-Time Reverse Transcription PCR
The total RNA was isolated using TRIzol agent (TransGen Biotech, Beijing, China), and each RNA sample was reverse-transcribed to complementary DNA (cDNA) by the PrimeScript RT Reagent Kit (Takara, Dalian, Liaoning Province, China). cDNA was used for quantitative real-time polymerase chain reaction (qRT-PCR) analysis. The sets of primer pairs of the two pathogens and of the
nitric oxide synthase genes are listed in
Table 1, and the primer pairs of cytokines can be found in Nang et al.’s paper [
23]. For qRT-PCR reactions, the 25 μL reaction mixture included 2 μL cDNA, 12.5 μL SYBR Premix Ex TaqTM II (Takara, Beijing, China), 1.0 μL of forward primer and 1.0 μL of reverse primer, and 8.5 μL RNAase-free water (Takara, Beijing, China). The reaction conditions were 95 °C for 3 min, followed by 44 cycles of 95 °C for 10 s, then the specific melting temperature (Tm) of a primer pair for 30 s, and then 95 °C for 10 s and 72 °C for 10 s, using a Bio-Rad IQ5 Thermal Cycler (Bio-Rad). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was selected as a reference gene. The expression fold changes were calculated using the 2
−△△Ct method [
24].
2.5. Determination of Cell Mortality
Cell mortality was detected by a CellTox Green Cytotoxicity Assay Kit (Promega, Wisconsin, United States). The detection system uses a proprietary asymmetric cyanine dye, which is blocked by living cells but can stain the DNA of dead cells. When the dye binds with DNA, it emits fluorescence. Therefore, the fluorescence signal produced by the dye bound to the dead cell DNA is directly proportional to the cytotoxicity. We operated strictly according to the protocol, adding 50 μL cells with a concentration of 1 × 105/mL to the 96-well cell culture plate. Then, we added 100 μL CellToxTM Green reagent (Madison, WI, USA), and after incubation for 15 min, it was put into a fluorescence enzyme labeling instrument. After 1 min of simple oscillation, the fluorescence signal value was detected at 485–500 nm Ex/520~530 nm Em wavelength.
2.6. Determination of iNOS Activity
The inducible nitric oxide synthase (iNOS) activity was detected by a Nitric Oxide Synthase (NOS) Activity Assay Kit (BioVision, SMB, Milpitas, CA, United States). Briefly, we added 100–200 μL cold NOS Assay Buffer containing protease inhibitor cocktail to fresh cells (2–5 × 106), which was then homogenized to disrupt the cells. The tissue or cell homogenate was centrifuged at 10,000× g and 4 °C for 10 min. The clarified supernatant was transferred to a fresh pre-chilled tube and kept on ice. The protein concentration was measured, and the lysates were used immediately to assay NOS activity. Then, 30–60 μL (200–400 μg protein) of cell/tissue homogenate or purified protein was measured into the desired wells in a 96-well plate. For the positive control, NOS enzyme was diluted 1:20 in NOS Dilution Buffer just before use. Next, 5–10 μL of the diluted NOS enzyme was added into the desired well(s), and the volume of the sample and the positive control wells were made up to 60 μL/well with the NOS Assay Buffer. Enough reaction mixes for the number of wells (standards, positive control, and sample) were prepared to be analyzed, which were then mixed well and incubated at 37 °C for 1 h. After incubation, 90 μL of NOS Assay Buffer was added to the standard, positive control, and sample wells, and subsequently 5 μL of the enhancer was added into each well, which were then mixed and incubated at room temperature for 10 min. After this, 50 μL of Griess Reagent 1 and 50 μL of Griess Reagent 2 were added to the standard, positive control, and sample wells, which were then mixed and incubated for 10 min, before the absorbance (540 nm) was read using a microplate reader. The nitrite standard curve was plotted and the iNOS activity calculated according to the formula: sample iNOS activity = nitrite amount in sample well from standard curve/(reaction time × amount of protein) = pmol/min/μg = mU/mg.
2.7. Nitrite Quantification
Nitrite levels were determined by colorimetric assay based on the Griess reaction (Beyotime, Haimen, Jiangsu Province, China), using sodium nitrite standards. Briefly, 100 μL of cell-free pretreated supernatant was mixed with 100 μL of Griess reagent, and after 10 min, the absorbance was measured at 540 nm wavelength. Using a standard curve, the absorbance of the samples was converted to micromolar nitric oxide (NO).
2.8. Detection of Cell Phagocytosis
The phagocytosis of macrophages was detected by a fluorescein-labeled Escherichia coli K-12 BioParticles of Vybrant Phagocytosis Assay Kit (ThermoFisher, MA, United States). Briefly, 100 µL of the prepared fluorescent BioParticle suspension was added to all the negative control, positive control, and experimental wells, and incubated for 2 h. The BioParticle was removed and 100 µL of the prepared trypan blue suspension was immediately added to all wells, before incubating for 1 min at room temperature. The experimental and control wells of the microplate were read in the fluorescence plate reader using ~480 nm excitation, ~520 nm emission, and the appropriate sensitivity settings. The net phagocytosis and the response to the phagocytosis effector agent were then calculated. First, the average fluorescence intensity of a group of negative control wells was subtracted from that of a group of positive control wells to yield the net positive reading. Second, the average fluorescence intensity of a group of negative-control wells was subtracted from that of a group of identical experimental wells to obtain the net experimental reading. The phagocytosis response to the effector could then be expressed as follows: % effect = net experimental reading/net positive reading × 100%.
2.9. mRNA Expression of Cytokines by RT-PCR and Quantitative Secretions by ELISA Kits
The total RNA was extracted from HD11 cells by applying Trizol (TransGen Biotech, Beijing, China), and was subsequently treated with a DNA-free kit to filter DNA contamination. Relative quantification of interleukin (IL)-1β, IL-2, IL-6, IL-10, Interferon gamma (IFN-γ) and tumor necrosis factor-α (TNF-α) was performed using an SYBR Green PCR Master Mix kit (Takara, Dalian, China), as previously described [
15]. As for cytokine determination, roughly 200 μL aliquots of each sample were used to measure the cytokines IL-1β, IL-2, IL-6, IL-10, IFN-γ, and TNF-α with commercial ELISA kits (Kingfisher Biotech Inc., Saint Paul, MN, United States).
2.10. Statistical Analysis
The data are presented as averages ± standard deviations (SDs), as indicated. Statistical comparisons were analyzed with one-way ANOVA with the Least-Significant Difference (LSD) post hoc test. All of the statistical analyses were performed with SPSS version 25.0. When p > 0.05, the results were not significant; when p < 0.05, the results were significantly different; when p < 0.01, the results were extremely significantly different.
4. Discussion
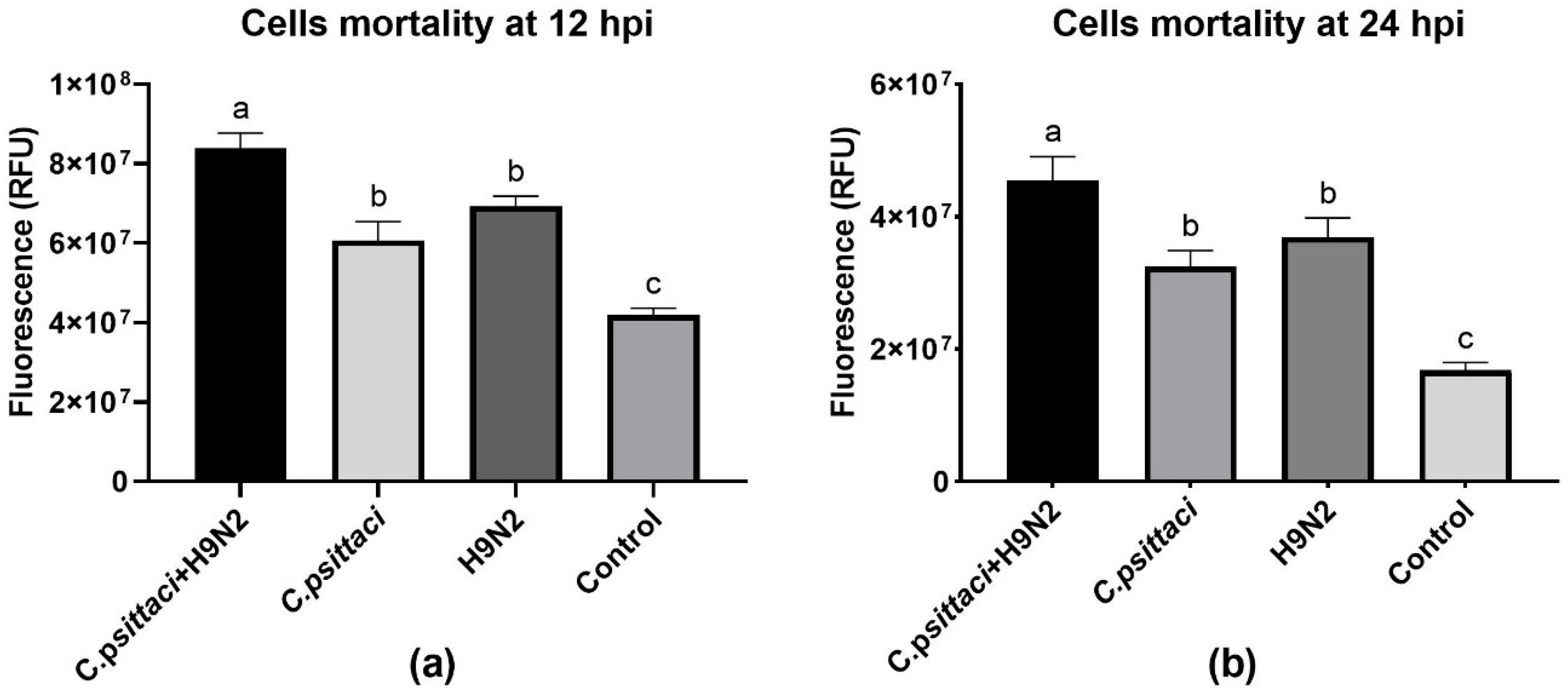
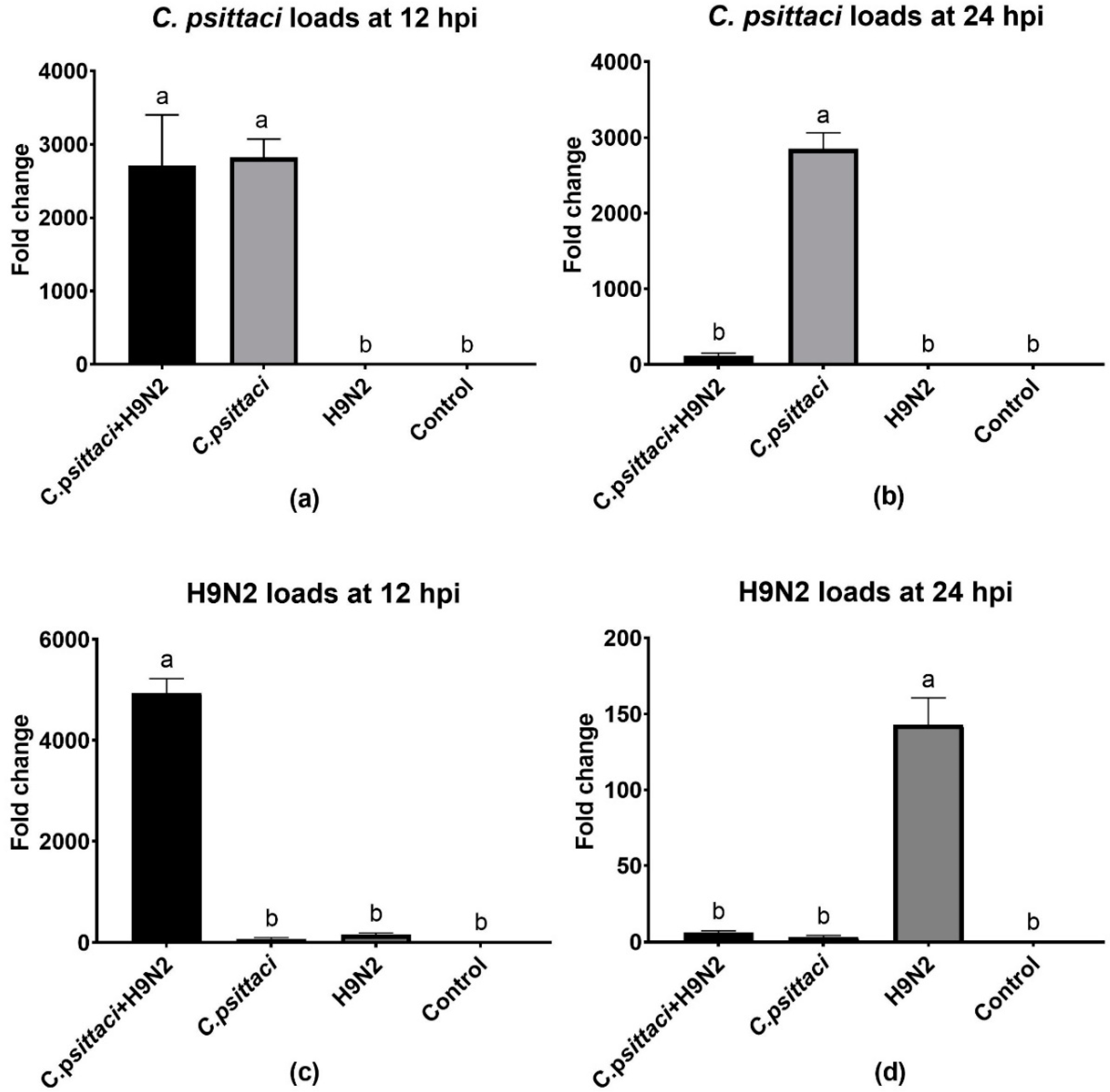
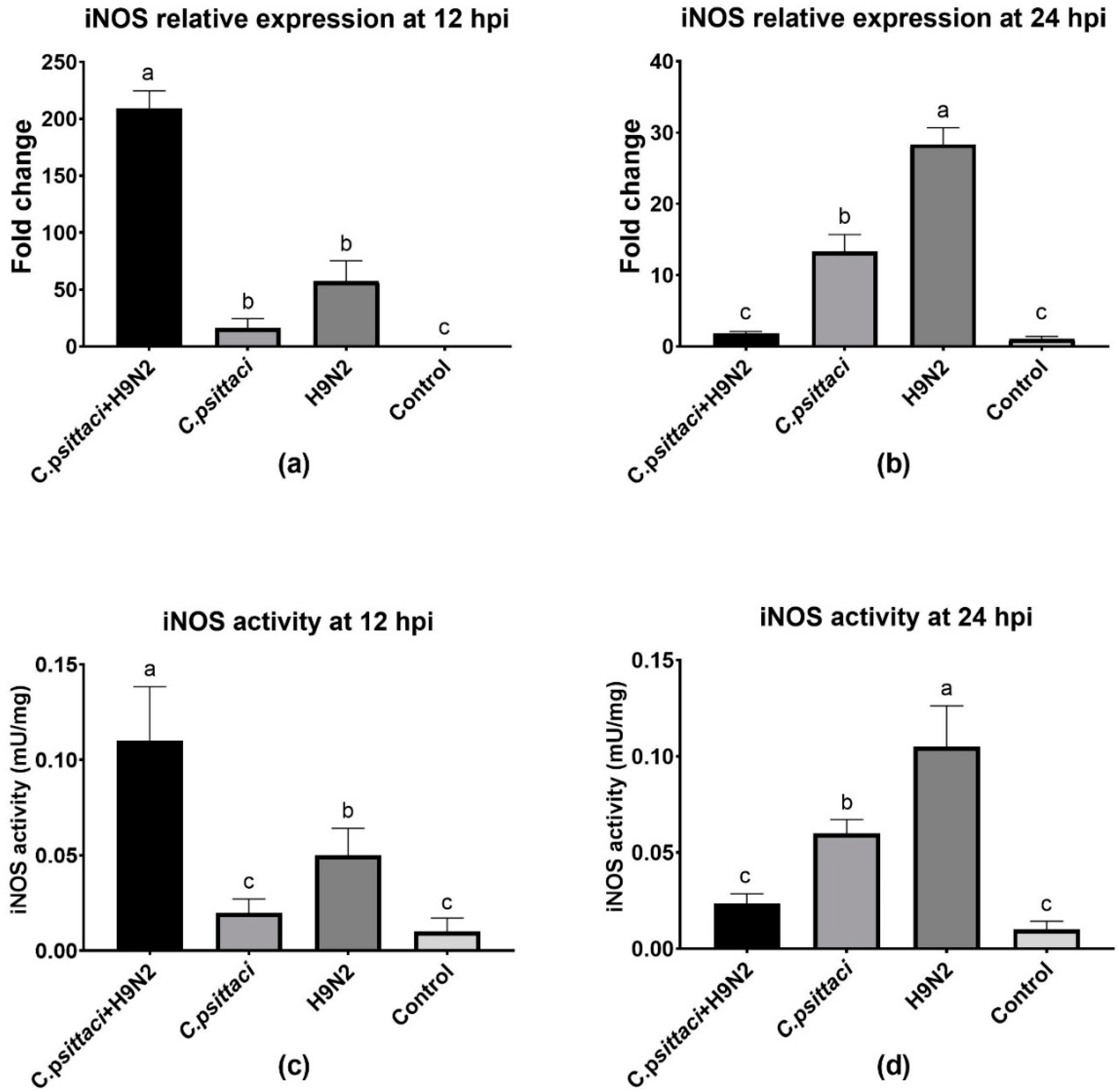
In the present study, we investigated the effect of chicken macrophage functions in response to coinfection with C. psittaci and H9N2 avian influenza virus. Our major finding was that coinfection with C. psittaci and H9N2 could significantly aggravate the mortality of HD11 cells compared to the effects of infection with C. psittaci or H9N2 alone. Moreover, the iNOS expression and enzyme activity, as well as NO concentration, of HD11 cells were significantly reduced compared to those of C. psittaci or H9N2 alone. In other words, coinfection of C. psittaci with H9N2 could stimulate HD11 cells to express less iNOS and NO compared to infection with H9N2 alone. In addition, coinfection of C. psittaci and H9N2 induced lower phagocytosis of HD11 cells than H9N2 or C. psittaci alone at 24 hpi. Furthermore, both the mRNA expression and cytokine levels of IL-4 and IL-10 were significantly increased at 24 hpi in the C. psittaci + H9N2 group. On the contrary, the Th1 cytokines of IL-1β, IL-2, and IL-6 were significantly increased in the C. psittaci + H9N2 group compared to those of C. psittaci or H9N2 alone at the early stage. Later, only TNF-α secretions were increased significantly compared to those of the C. psittaci group at 24 hpi. All the above data support our hypothesis that C. psittaci infection could aggravate the infection of H9N2 by impairing macrophage functions, leading to the outbreak of severe respiratory diseases.
In our previous study, we established an SPF chicken animal model with coinfection of
C. psittaci and H9N2, and we found that
C. psittaci infection increased the mortality of H9N2 by inhibiting humoral immunity and cellular immunity, as well as by altering the Th1/Th2 balance, ultimately weakening the immune system of the body [
15]. It was the first report that
C. psittaci infection can induce immune suppression in vivo and can lead to increased susceptibility to H9N2 infection. It also suggested that we should consider primary infection by
C. psittaci in any respiratory disease, and should eradicate it during the treatment of avian respiratory disease. In addition, other studies have also reported the role of
C. psittaci and H9N2 in the pathogenesis of coinfection. In this sense, coinfection is a common infection of two or more pathogens in the same host. Similar coinfection occurs frequently in human cases. For example, in 1918, in the human influenza virus pandemic, almost all cases of death were caused by bacteria mixed infection, and the additional bacterial infection greatly increased the risk of death [
25]. Moreover, the combination of malaria and helminth is prevalent in less-developed countries [
26]. Hence, identifying the primary pathogen of the mixed infection has been underestimated due to limited investigation. Traditionally, low pathogenic avian influenza virus triggers primary infection, and then the secondary infection of bacteria follows [
27]. Since 2007, the outbreak of avian airsacculitis has been documented by the mixed infection of various pathogens [
28].
In our study, coinfection with
C. psittaci and H9N2 increased the mortality of HD11 cells in comparison with
C. psittaci or H9N2 alone. Simultaneous infection with
C. psittaci and H9N2 was able to increase the mortality of HD11 cells by decreasing iNOS activity and phagocytosis, indicating that coinfection might impair macrophage functions and facilitate the immune evasions of the two pathogens. Our discoveries are consistent with a previous report that showed macrophage functions were damaged post-infection with virulent
C. psittaci [
19].
Macrophages play an important role in innate immunity, such as phagocytosis, antiviral infection, and enhanced immune regulation. Pathogens infect the body and the monocytes after infection of the main target cells. After entering macrophages,
Chlamydia can escape immune surveillance and transport to the whole body with macrophages, and thus macrophages act as the delivery system for
Chlamydia [
19]. Therefore, it is necessary to study how macrophages play a role in the host’s immune response to the pathogen, as well as the pathogenesis of the pathogen, after the study of
Chlamydia and H9N2 infection. The
inducible nitric oxide synthase (
iNOS) gene and its activity have been shown to be involved in the reaction of L-arginine decomposition into NO and L-citrulline. Endotoxin or cytokines, such as LPS and IFN-γ, can induce chicken macrophages to produce iNOS, and to further produce NO [
29]. Our research shows that coinfection with
C. psittaci and H9N2 could significantly decrease the iNOS expression level and enzyme activity, as well as the NO concentration, of HD11 cells compared to H9N2 or
C. psittaci infection alone. However, the potential mechanism is unknown. The monocyte macrophage system has the function of phagocytosis and the killing of pathogens and tumor cells directly. In our study, we found that coinfection induced lower phagocytosis of HD11 cells compared to H9N2 infection. All this evidence illustrates the mechanism of macrophage dysfunction post-coinfection or after
C. psittaci infection alone. Cytokines secreted by macrophages are also important components of their immune regulation. Here, several pro- and anti-inflammatory cytokines were determined by mRNA expression and ELISA cytokine kits. The role of Th1/Th2 cell cytokines is important in the immune response to chlamydial infection. It appears that the Th1 CD4 cell response plays a dominant role in protective immunity, while Th2 CD4 cytokines (particularly IL-10) play a role in the immunopathology of chlamydial infection. Products of Th2 cells do not facilitate the production of IFN-γ or inhibit
Chlamydia growth [
30]. TNF-α regulates critical cell functions, including cell proliferation, survival, differentiation, and apoptosis. Macrophages are the major producers of TNF-α, and TNF-α has been shown to play a pivotal role in orchestrating the cytokine cascade in many inflammatory diseases [
31]. Increased TNF-α levels have been associated with atherosclerosis and coronary heart disease post-infection with
Chlamydia pneumoniae (
C. pneumoniae) [
32]. IL-10 is an anti-inflammatory cytokine known to play a critical role in chronic infections caused by intra-cellular organisms. During chlamydial infection, high IL-10 secretion was shown to be associated with pathogenesis in a mouse model of
Chlamydia trachomatis (
C. trachomatis) infection [
33]. In the present study, high TNF-α production was induced by coinfection with
C. psittaci and H9N2, and this might be associated with increasing the increased mortality of the HD11 cells. In our results, the expression of IL-4 and IL-10 were increased significantly in the coinfection groups, suggesting that chlamydial infection mediated the polarization of Th1/Th2 to the Th2 direction. The dominant role of Th2 cytokines hampers immune function, so that pathogens can evade immune surveillance and immune attacks. The Th2 polarization was consistent with our previous data, implying that immune suppression is aggravated post-artificial infection with H9N2 and
C. psittaci [
15].
In conclusion, coinfection often increases disease severity in both humans and animals. Understanding the mechanisms and effects of coinfection will improve the understanding of the mechanism of mixed infection. In this study, the macrophage functions were exacerbated by reducing phagocytosis post-coinfection. Also, the polarization of IL-4 and IL-10 expressions might contribute to macrophage dysfunction and facilitate H9N2 circulations by reducing virus clearance in host cells. In addition, further findings of this study show that not only does C. psittaci aggravate H9N2 infection, but H9N2 can also increase the mortality caused by C. psittaci infection. Whether this is beneficial to the infection and spread of C. psittaci remains to be confirmed. These findings suggest that we should consider the primary and latent infection of C. psittaci in respiratory disease and should eradicate C. psittaci during treatment.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}