The Effects of Different Concentrate-to-Forage Ratio Diets on Rumen Bacterial Microbiota and the Structures of Holstein Cows during the Feeding Cycle

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals, Diets and Sample Collection
2.3. Ruminal Fermentation Parameters
2.4. DNA Extraction and 16S rRNA Gene Amplicon Preparation
2.5. Illumina HiSeq Sequencing and Sequence Analysis
2.6. Bioinformatics and Statistical Analysis
3. Results
3.1. Dynamic Changes of Rumen Fermentation Characteristics of Cows Fed Two Different Diets
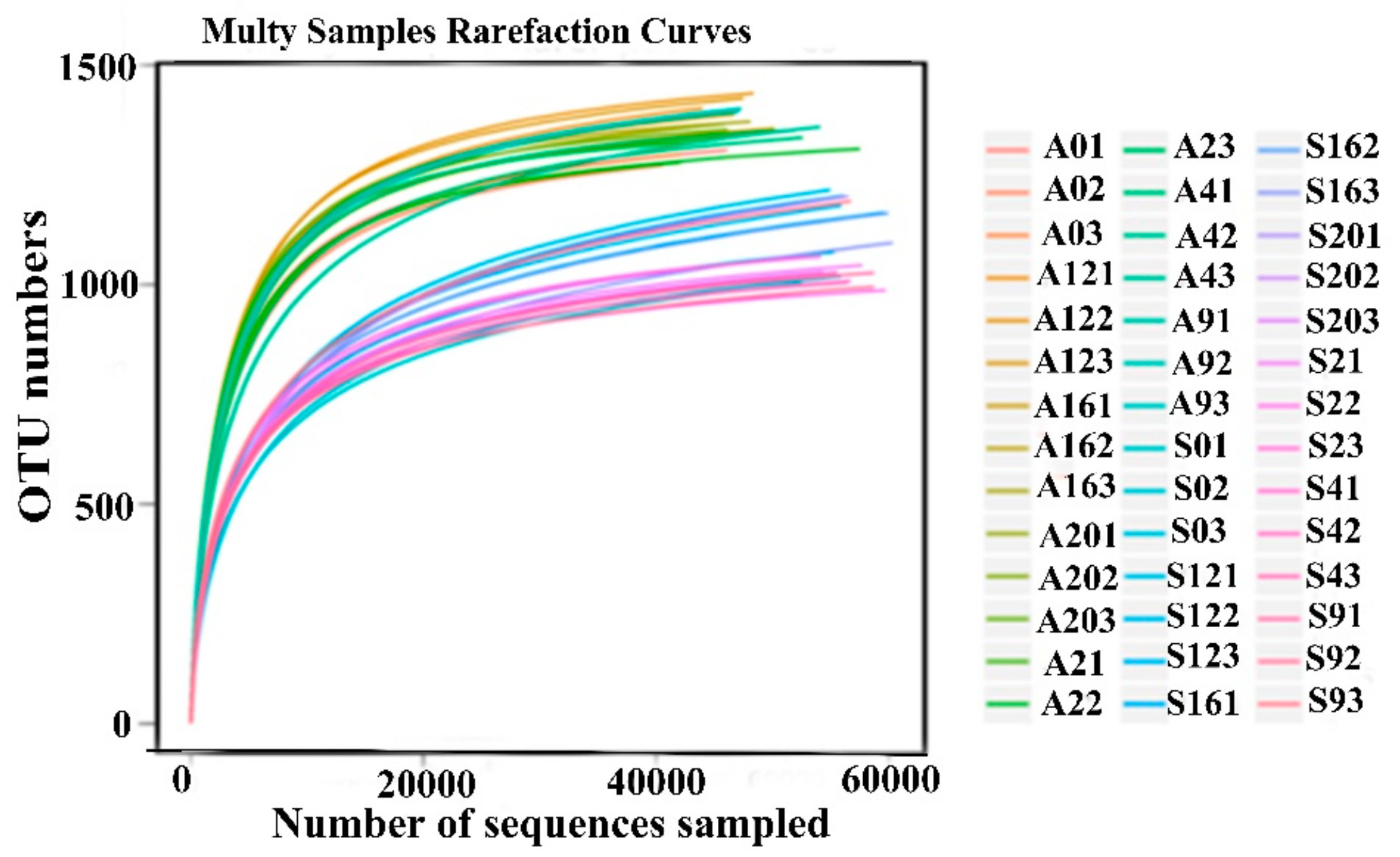
3.2. Sequencing Depth and Rumen Bacteria Diversity of Cows Fed Two Different Diets
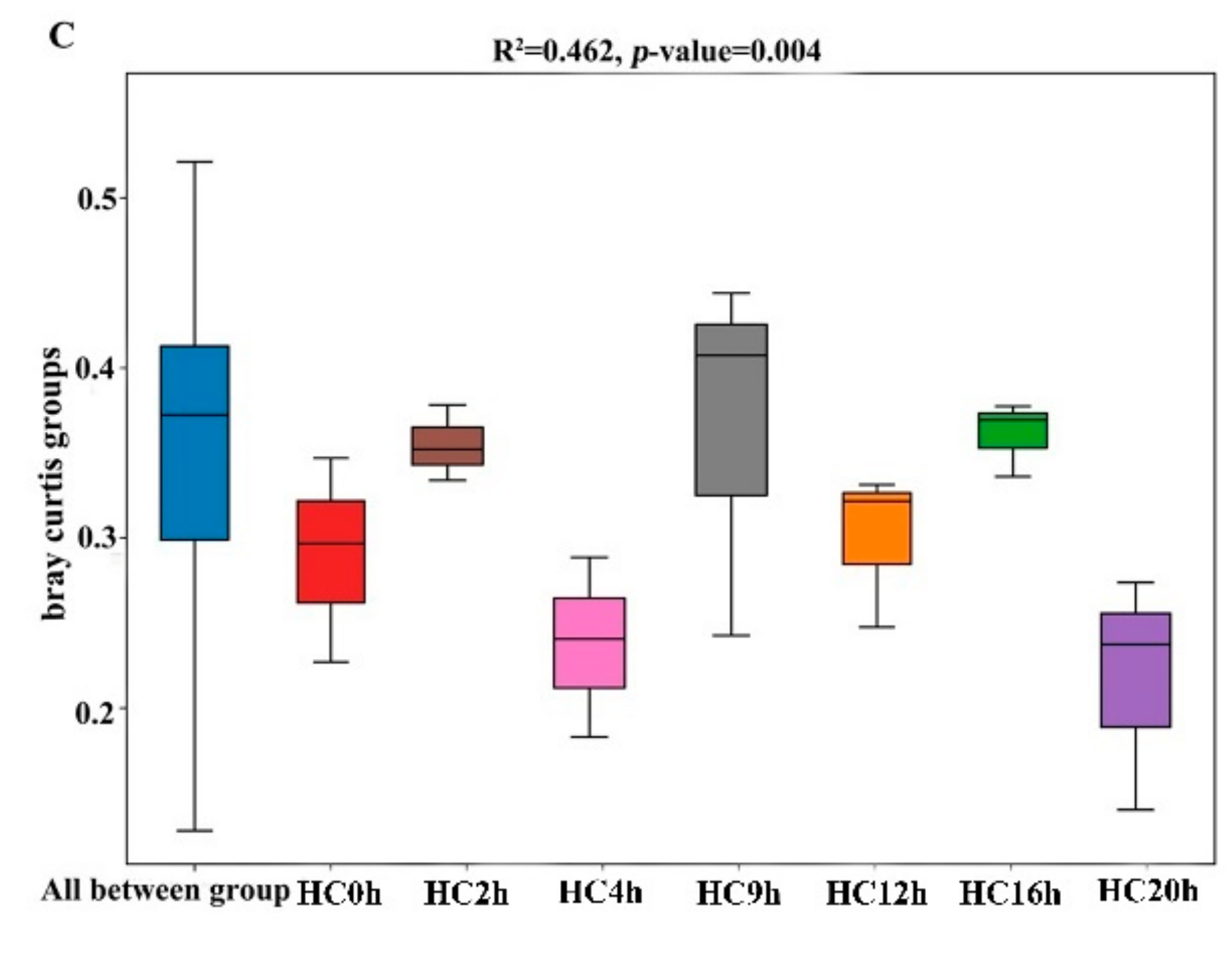
3.3. Ruminal Bacteria Changes within the Two Treatments During the Feeding Cycle
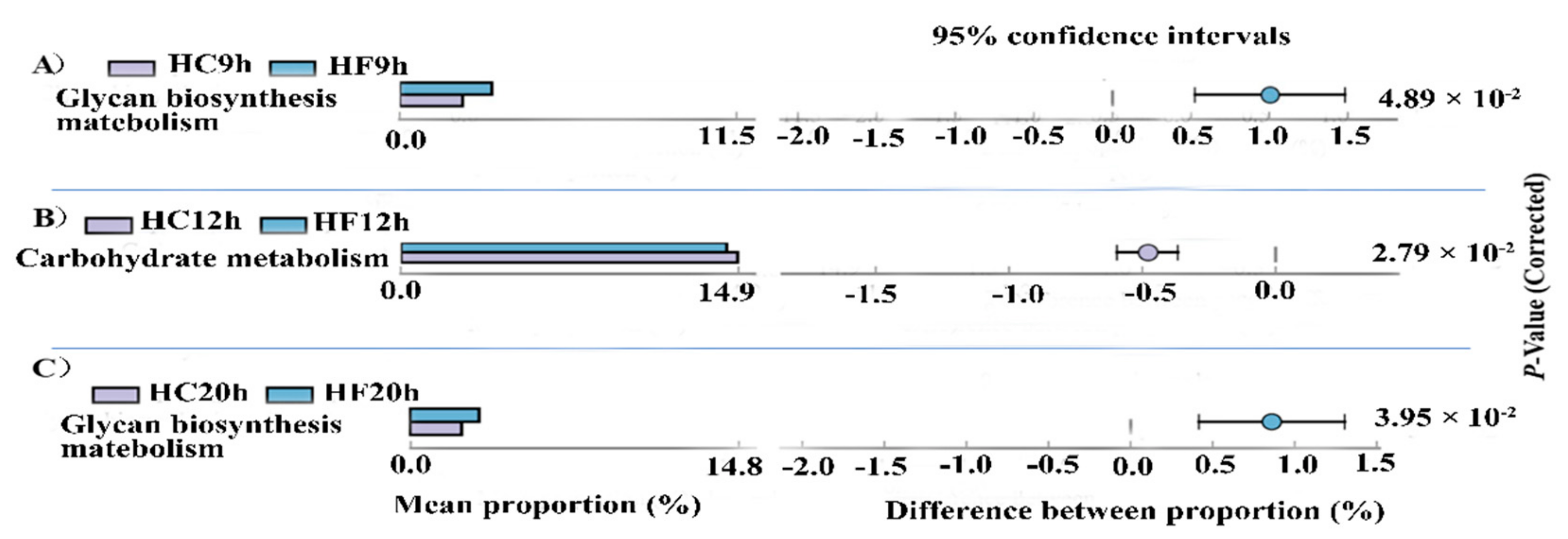
3.4. The Difference in the Microbial Composition Analysis and Functional Gene Prediction between the HF and HC Dietary Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Mao, S.; Zhang, M.; Liu, J.; Zhu, W. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci. Rep. 2015, 5, 16116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hespell, R.B.; Akin, D.E.; Dehority, B.A.; Mackie, R.I.; White, B.A. Bacteria, fungi and protozoa of the rumen. Gastrointest. Microbiol. 1997, 2, 325–332. [Google Scholar]
- Lin, B.; Henderson, G.; Zou, C.; Cox, F.; Liang, X.; Janssen, P.H. Characterization of the rumen microbial community composition of buffalo breeds consuming diets typical of dairy production systems in Southern China. Anim. Feed Sci. Tech. 2015, 207, 75–84. [Google Scholar] [CrossRef]
- Moraïs, S.; Mizrahi, I. The Road Not Taken: The Rumen Microbiome, Functional Groups, and Community States. Trends Microbiol. 2019, 27, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, B.; Wang, X.; Chen, Y.; Yang, Y. Effect of dietary concentrate to forage ratios on ruminal bacterial and anaerobic fungal populations of cashmere goats. Anaerobe 2019, 59, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, A.; Pacheco, D.; Hoskin, S.O.; Luo, D. Sheep Rumen Fermentation Characteristics Affected by Feeding Frequency and Feeding Level When Fed Fresh Forage. Animals 2020, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Teng, Z.; Lang, C.; Zhou, H.; Zhong, W.; Ban, Z. Effect of different forage-to-concentrate ratios on ruminal bacterial structure and real-time methane production in sheep. PLoS ONE 2019, 14, e0214777. [Google Scholar] [CrossRef]
- Ingvartsen, K.L. Feeding- and management-related diseases in the transition cow: Physiological adaptations around calving and strategies to reduce feeding-related diseases. Anim. Feed Sci. Tech. 2006, 126, 175–213. [Google Scholar] [CrossRef]
- Papi, N.; Mostafa, T.A.; Amanlou, H.; Memarian, M. Effects of dietary forage-to-concentrate ratios on performance and carcass characteristics of growing fat-tailed lambs. Anim. Feed Sci. Tech. 2011, 163, 93–98. [Google Scholar] [CrossRef]
- Machado, S.C.; Mcmanus, C.M.; Stumpf, M.T.; Fischer, V. Concentrate: forage ratio in the diet of dairy cows does not alter milk physical attributes. Trop. Anim. Health Prod. 2014, 46, 855–859. [Google Scholar] [CrossRef]
- Mialon, M.M.; Martin, C.; Garcia, F.; Menassol, J.B.; Dubroeucq, H.; Veissier, I. Effects of the forage-to-concentrate ratio of the diet on feeding behaviour in young Blond d’Aquitaine bulls. Animal 2008, 2, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Grilli, D.J.; Fliegerová, K.; Kopečný, J.; Lama, S.P.; Egea, V.; Sohaefer, N. Analysis of the rumen bacterial diversity of goats during shift from forage to concentrate diet. Anaerobe 2016, 42, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, H.; Yang, P.; Zhao, J.; Huang, H.; Xue, X. Comparative quantitative analysis of gene expression profiles of glycoside hydrolase family 10 xylanases in the sheep rumen during a feeding cycle. Appl. Environ. Microbiol. 2013, 79, 1212–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kljak, K.; Pino, F.; Heinrichs, A.J. Effect of forage to concentrate ratio with sorghum silage as a source of forage on rumen fermentation, N balance, and purine derivative excretion in limit-fed dairy heifers. J. Dairy Sci. 2017, 100, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welkie, D.G.; Stevenson, D.M.; Weimer, P.J. ARISA analysis of ruminal bacterial community dynamics in lactating dairy cows during the feeding cycle. Anaerobe 2010, 16, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.S.; Duncan, S.H. The effect of avoparcin on cellulolytic bacteria of the ovine rumen. J. Gen. Microbiol. 1985, 131, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.L.; Blum, R.; Glennon, W.E.; Burton, A.L. Measurement of carboxymethylcellulase activity. AnBio 1960, 1, 127–132. [Google Scholar] [CrossRef]
- Minas, K.; Mcewan, N.R.; Newbold, C.J.; Scott, K.P. Optimization of a high-throughput CTAB-based protocol for the extraction of qPCR-grade DNA from rumen fluid, plant and bacterial pure cultures. FEMS Microbiol Lett. 2011, 325, 162–169. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Dong, X.; Dong, Z. Prokaryote diversity in the rumen of yak (Bos grunniens) and Jinnan cattle (Bos taurus) estimated by 16S rDNA homology analyses. Anaerobe 2005, 11, 207–215. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH fast length adjustment of short reads to improve genome assemblies. In Bioinformatics; 2010; Volume 27, pp. 2957–2963. [Google Scholar]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski., J.; Stombaugh., J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A language and environment for statistical computing. Computing 2014, 14, 12–21. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Ash, R.W.; Dobson, A. The effect of absorption on the acidity of rumen contents. J. Physiol. 1963, 169, 39–61. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shi, H.T.; Wang, Y.J.; Li, S.L.; Cao, Z.J.; Ji, S.K.; He, Y.; Zhang, H.T. Effect of dietary forage to concentrate ratios on dynamic profile changes and interactions of ruminal microbiota and metabolites in holstein heifers. Front Microbiol. 2017, 8, 2206–2224. [Google Scholar] [CrossRef] [Green Version]
- Hristov, A.N.; Zaman, S.; Schneider, C. Effect of dietary concentrate on rumen fermentation, digestibility, and nitrogen losses in dairy cows. J. Dairy Sci. 2010, 93, 4211–4222. [Google Scholar]
- Doyle, P.T.; Dove, H.; Freer, M.; Hart, F.J.; Dixon, R.M.; Egan, A.R. Effects of a concentrate supplement on the intake and digestion of a low-quality forage by lambs. J. Agric. Sci. 1988, 111, 503–511. [Google Scholar] [CrossRef]
- Grubb, J.A.; Dehority, B.A. Effects of an abrupt change in ration from all roughage to high concentrate upon rumen microbial numbers in sheep. Appl. Microbiol. 1975, 30, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaxter, K.L. The Energy Metabolism Of Ruminants. In Digestive Physiology and Metabolism in Ruminants: Proceedings of the 5th International Symposium on Ruminant Physiology, held at Clermont — Ferrand, on 3rd–7th September, 1979; Springer: Springfield, UK, 1962. [Google Scholar]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Collaborators, G.R.C.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef] [PubMed]
- Shabat, S.K.; Sasson, G.; Doron-Faigenboim, A.D.; Durman, T.; Yaacoby, S.; Berg-Miller, M.E.; White, B.A.; Shterzer, N.; Mizrahi, I. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef] [Green Version]
- Ellison, M.J.; Conant, G.C.; Cockrum, R.R.; Austin, K.J.; Truong, H.; Becchi, M.; Lamberson, W.R.; Cammack, K.M. Diet alters both the structure and taxonomy of the ovine gut microbial ecosystem. DNA Res. 2014, 21, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, R.M.; Schwaiger, T.; Penner, G.B.; Beauchemin, K.A.; . Forster, R.J.; McKinnon, J.J.; McAllister, T.A. Characterization of the core rumen microbiome in cattle during transition from forage to concentrate as well as during and after an acidotic challenge. PLoS ONE 2013, 8, e83424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, S.C.; Nd, P.H.; Najar, F.Z.; Sukharnikov, L.O.; Krehbiel, C.R.; Nagaraja, T.G.; Roe, B.A.; Desilva, U. Rumen Microbial Population Dynamics during Adaptation to a High-Grain Diet. Appl. Environ. Microbiol. 2010, 76, 7482–7490. [Google Scholar] [CrossRef] [Green Version]
- Bekele, A.Z.; Satoshi, K.; Yasuo, K. Genetic diversity and diet specificity of ruminal Prevotella revealed by 16S rRNA gene-based analysis. FEMS Microbiol. Lett. 2010, 305, 49–57. [Google Scholar] [CrossRef]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. Isme J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [Green Version]
- Hespell, R.B. The Genera Succinivibrio and Succinimonas. The Prokaryotes, Third ed.; Springer: Springfield, UK, 1992; pp. 3979–3982. [Google Scholar]
- Xue, M.Y.; Sun, H.Z.; Wu, X.H.; Guan, L.L.; Liu, J.X. Assessment of rumen bacteria in dairy cows with varied milk protein yield. J. Dairy Sci. 2019, 102, 5031–5041. [Google Scholar] [CrossRef]
- Plaizier, J.C.; Li, S.; Danscher, A.M.; Derakshani, H.; Andersen, P.H.; Khafipour, E. Changes in microbiota in rumen digesta and feces due to a grain-based subacute ruminal acidosis (SARA) challenge. MicEc 2017, 74, 485–495. [Google Scholar] [CrossRef]
- Han, K.N.; Jin, W.; Mao, Z.; Dong, S.; Zhang, Q.; Yang, Y.; Chen, B.; Wu, H.; Zeng, M. Microbiome and butyrate production are altered in the gut of rats fed a glycated fish protein diet. J. Funct. Foods 2018, 47, 423–433. [Google Scholar] [CrossRef]
- Iwamoto, M.; Asanuma, N.; Hino, T. Ability of Selenomonas ruminantium, Veillonella parvula, and Wolinella succinogenes to Reduce Nitrate and Nitrite with Special Reference to the Suppression of Ruminal Methanogenesis. Anaerobe 2002, 8, 209–215. [Google Scholar] [CrossRef]
- Mikx, F.H.M.; Hoeven, J.S. Symbiosis of Streptococcus mutans and Veillonella alcalescens in mixed continuous cultures. Arch. Oral Biol. 1975, 20, 407–410. [Google Scholar] [CrossRef]
- Ng, S.K.; Hamilton, I.R. Lactate metabolism by Veillonella parvula. J. Bacteriol. 1971, 105, 999–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, R.; Obura, M.; Odongo, D.; Odenyo, A. Specific PCR assay for a tannin-tolerant Selenomonas ruminantium isolate, derived from helicase coding sequences. Appl. Environ. Microbiol. 2004, 70, 3180–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, M.P. The characteristics of strains of Selenomonas isolated from bovine rumen contents. J. Bacteriol. 1956, 72, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.S.; Flint, H.J.; Bryant, M.P. The Rumen Bacteria. The Rumen Microbial Ecosystem, 2nd ed.; Springer: Springfield, UK, 1997; pp. 10–72. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HF | HC | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Time | Observed pH | C (H+) | Observed pH | C (H+) | |||||||||
| 0 h | 6.79 | 6.92 | 6.78 | 1.62 × 10−7 | 1.20 × 10−7 | 1.66 × 10−7 | 6.94 | 6.9 | 6.85 | 1.15 × 10−7 | 1.26 × 10−7 | 1.41 × 10−7 | |
| 2 h | 6.52 | 6.52 | 6.43 | 3.02 × 10−7 | 3.02 × 10−7 | 3.71 × 10−7 | 6.54 | 6.48 | 6.35 | 2.88 × 10−7 | 3.31 × 10−7 | 4.47 × 10−7 | |
| 4 h | 6.1 | 6.02 | 6.15 | 7.94 × 10−7 | 9.55 × 10−7 | 7.08 × 10−7 | 5.85 | 5.61 | 5.73 | 1.41 × 10−6 | 2.45 × 10−6 | 1.86 × 10−6 | |
| 6 h | 6.38 | 6.12 | 6.28 | 4.17 × 10−7 | 7.58 × 10−7 | 5.25 × 10−7 | 5.61 | 5.5 | 5.62 | 2.45 × 10−6 | 3.16 × 10−6 | 2.40 × 10−6 | |
| 9 h | 6.47 | 6.17 | 6.31 | 3.39 × 10−7 | 6.76 × 10−7 | 4.90 × 10−7 | 5.88 | 5.64 | 5.74 | 1.32 × 10−6 | 2.29 × 10−6 | 1.82 × 10−6 | |
| 12 h | 6.53 | 6.31 | 6.41 | 2.95 × 10−7 | 4.90 × 10−7 | 3.89 × 10−7 | 6.12 | 6.07 | 5.94 | 7.59 × 10−7 | 8.51 × 10−7 | 1.15 × 10−6 | |
| 16 h | 6.69 | 6.6 | 6.65 | 2.04 × 10−7 | 2.51 × 10−7 | 2.24 × 10−7 | 6.43 | 6.39 | 6.35 | 3.72 × 10−7 | 4.07 × 10−7 | 4.47 × 10−7 | |
| 20 h | 6.79 | 6.68 | 6.68 | 1.62 × 10−7 | 2.09 × 10−7 | 2.09 × 10−7 | 6.58 | 6.61 | 6.55 | 2.63 × 10−7 | 2.45 × 10−7 | 2.82 × 10−7 | |
| 24 h | 6.92 | 6.78 | 6.79 | 1.20 × 10−7 | 1.66 × 10−7 | 1.62 × 10−7 | 6.88 | 6.83 | 6.74 | 1.32 × 10−7 | 1.48 × 10−7 | 1.82 × 10−7 | |
| Item | Diet | Time | SEM | p-Values | |||||||||
| 0 h | 2 h | 4 h | 9 h | 12 h | 16 h | 20 h | 24 h | Feed | Time | Feed * Time | |||
| C(H+) | HF | 1.50 × 10−7 e | 3.25 × 10−7 cd | 8.19 × 10−7 Ba | 5.02 × 10−7 Bb | 3.91 × 10−7 Bbc | 2.26 × 10−7 Bde | 1.93 × 10−7 Bde | 1.50 × 10−7 e | 0.000 | <0.0001 | <0.0001 | <0.0001 |
| HC | 1.27 × 10−7 c | 3.55 × 10−7 c | 1.91 × 10−6 Aa | 1.81 × 10−6 Aa | 9.19 × 10−7 Ac | 4.09 × 10−7 Ac | 2.63 × 10−7 Ac | 1.54 × 10−7 c | |||||
| pH | HF | 6.83 | 6.49 | 6.09 | 6.30 | 6.41 | 6.65 | 6.71 | 6.83 | ||||
| HC | 6.90 | 6.45 | 5.72 | 5.74 | 6.04 | 6.39 | 6.58 | 6.81 | |||||
| Item | Diet | After Feed Time (h) | SEM | p-Values | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 h | 2 h | 4 h | 9 h | 12 h | 16 h | 20 h | Feed | Time | Feed * Time | |||
| TVFA a, mM | HF | 106.74 d | 125.88 cd | 155.38 a | 147.09 ab | 128.65 bc | 110.29 cd | 116.83 cd | 6.5148 | 0.167 | <0.001 | 0.174 |
| HC | 118.35 b | 137.47 b | 164.60 a | 159.40 a | 135.67 b | 133.04 b | 125.57 b | |||||
| VFA b, molar % of TVFA | ||||||||||||
| Acetate | HF | 69.75 Abc | 70.69 Abc | 73.65 Aab | 76.38 Aa | 71.96 Ab | 67.20 Ac | 67.54 Ac | 0.770 | <0.001 | <0.001 | <0.001 |
| HC | 61.54 Ba | 57.46 Bb | 57.28 Bb | 53.73 Bd | 54.80 Bcd | 54.54 Bcd | 56.25 Bbc | |||||
| Propionate | HF | 16.09 Babc | 16.35 Babc | 14.39 Bc | 14.47 Bbc | 17.08 Bab | 18.37 Ba | 18.48 Ba | 0.576 | <0.001 | <0.001 | <0.001 |
| HC | 24.98 Ad | 27.86 Abc | 27.66 Ac | 29.59 Aa | 29.54 Aa | 29.03 Aa | 28.82 Aab | |||||
| Butyrate | HF | 7.82 Bc | 7.83 Bc | 8.04 Bbc | 5.84 Bd | 7.50 Bc | 10.16 Ba | 9.112 Bab | 0.557 | <0.001 | 0.001 | <0.001 |
| HC | 9.17 Ad | 10.35 Ad | 11.19 Ac | 13.17 Aa | 12.51 Ab | 12.56 Ab | 11.13 Ac | |||||
| Isobutyrate | HF | 4.85 Aa | 3.75 Ab | 2.89 Ade | 2.42 Ae | 2.50 Ae | 3.19 Acd | 3.65 Abc | 0.274 | <0.001 | <0.001 | 0.008 |
| HC | 1.09 Ba | 0.94 Bb | 0.81 Bc | 0.72 Bd | 0.69 Bd | 0.86 Bc | 0.88 Bbc | |||||
| Valerate | HF | 0.50 Bb | 0.62 Ba | 0.46 Bb | 0.40 Bb | 0.46 Bb | 0.45 Bb | 0.44 Bb | 0.054 | <0.001 | <0.001 | 0.090 |
| HC | 1.74 Ab | 1.91 Aa | 1.78 Aab | 1.56 Acd | 1.49 Ad | 1.79 Aab | 1.66 Abc | |||||
| Isovalerate | HF | 1.00 Ba | 0.76 Bb | 0.56 Bcd | 0.49 Bd | 0.50 Bd | 0.64 Bbc | 0.76 Bb | 0.049 | <0.001 | <0.001 | 0.018 |
| HC | 1.48 Aa | 1.46 Aa | 1.27 Ab | 1.22 Ab | 0.97 Ac | 1.21 Ab | 1.26 Ab | |||||
| A:P c | HF | 4.4 Ab | 4.41 Ab | 5.12 Aa | 5.31 Aa | 4.22 Abc | 3.69 Ac | 3.66 Ac | 0.172 | <0.001 | <0.001 | <0.001 |
| HC | 2.46 Ba | 2.06 Bb | 2.07 Bb | 1.82 Bd | 1.85 Bcd | 1.88 Bcd | 1.95 Bbc | |||||
| Sample | After Feed Time (h) | SEM | p-Values | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 h | 2 h | 4 h | 9 h | 12 h | 16 h | 20 h | Feed | Time | Feed * Time | |||
| OTU | HF | 1291 Ad | 1304 Ad | 1348 Ac | 1396 Aab | 1420 Aa | 1371 Abc | 1353 Ac | 18.19 | <0.001 | <0.001 | 0.228 |
| HC | 1032 Bc | 1031 Bc | 1013 Bc | 1070 Bbc | 1186 Ba | 1153 Bab | 1097 Babc | |||||
| ACE | HF | 1116 bc | 1106 c | 1131 Aabc | 1147 ab | 1169 Ba | 1139 Babc | 1133 Babc | 19.59 | <0.001 | 0.003 | 0.174 |
| HC | 1110 c | 1083 c | 1079 Bc | 1153 bc | 1327 Aa | 1294 Aab | 1190 Aabc | |||||
| Chao 1 | HF | 1126 bc | 1112 c | 1145 Abc | 1157 ab | 1193 Ba | 1147 Bbc | 1142 bc | 21.04 | <0.001 | 0.012 | 0.223 |
| HC | 1131 c | 1094 c | 1093 Bc | 1177 bc | 1377 Aa | 1331 Aab | 1222 abc | |||||
| Shannon | HF | 5.51 Abc | 5.55 abc | 5.45 Ac | 5.52 bc | 5.66 Aa | 5.59 Aab | 5.66 Aa | 0.07 | <0.001 | 0.315 | 0.273 |
| HC | 4.88 Bb | 5.23 ab | 5.01 Bab | 5.34 a | 5.05 Bab | 5.23 Bab | 4.90 Bb | |||||
| Simpson | HF | 0.011 Bab | 0.011 Bab | 0.011 Ba | 0.011 ab | 0.009 Bbc | 0.010 abc | 0.009 Bc | 0.0061 | <0.001 | 0.018 | 0.600 |
| HC | 0.030 A | 0.033 A | 0.029 A | 0.016 | 0.026 A | 0.021 | 0.035 A | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Li, Y.; Zhang, Y.; Wang, L. The Effects of Different Concentrate-to-Forage Ratio Diets on Rumen Bacterial Microbiota and the Structures of Holstein Cows during the Feeding Cycle. Animals 2020, 10, 957. https://doi.org/10.3390/ani10060957
Wang L, Li Y, Zhang Y, Wang L. The Effects of Different Concentrate-to-Forage Ratio Diets on Rumen Bacterial Microbiota and the Structures of Holstein Cows during the Feeding Cycle. Animals. 2020; 10(6):957. https://doi.org/10.3390/ani10060957
Chicago/Turabian StyleWang, Lijun, Yang Li, Yonggen Zhang, and Lihua Wang. 2020. "The Effects of Different Concentrate-to-Forage Ratio Diets on Rumen Bacterial Microbiota and the Structures of Holstein Cows during the Feeding Cycle" Animals 10, no. 6: 957. https://doi.org/10.3390/ani10060957