Identification of the Key microRNAs and miRNA-mRNA Interaction Networks during the Ovarian Development of Hens


 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection, RNA Isolation, and Quality Analysis
2.3. Library Preparation and Sequencing
2.4. Distribution and Identification of miRNA
2.5. Differential Expression of miRNA and mRNA and Clustering Analysis
2.6. Target Gene Prediction, Enrichment, and Interaction Network Analysis
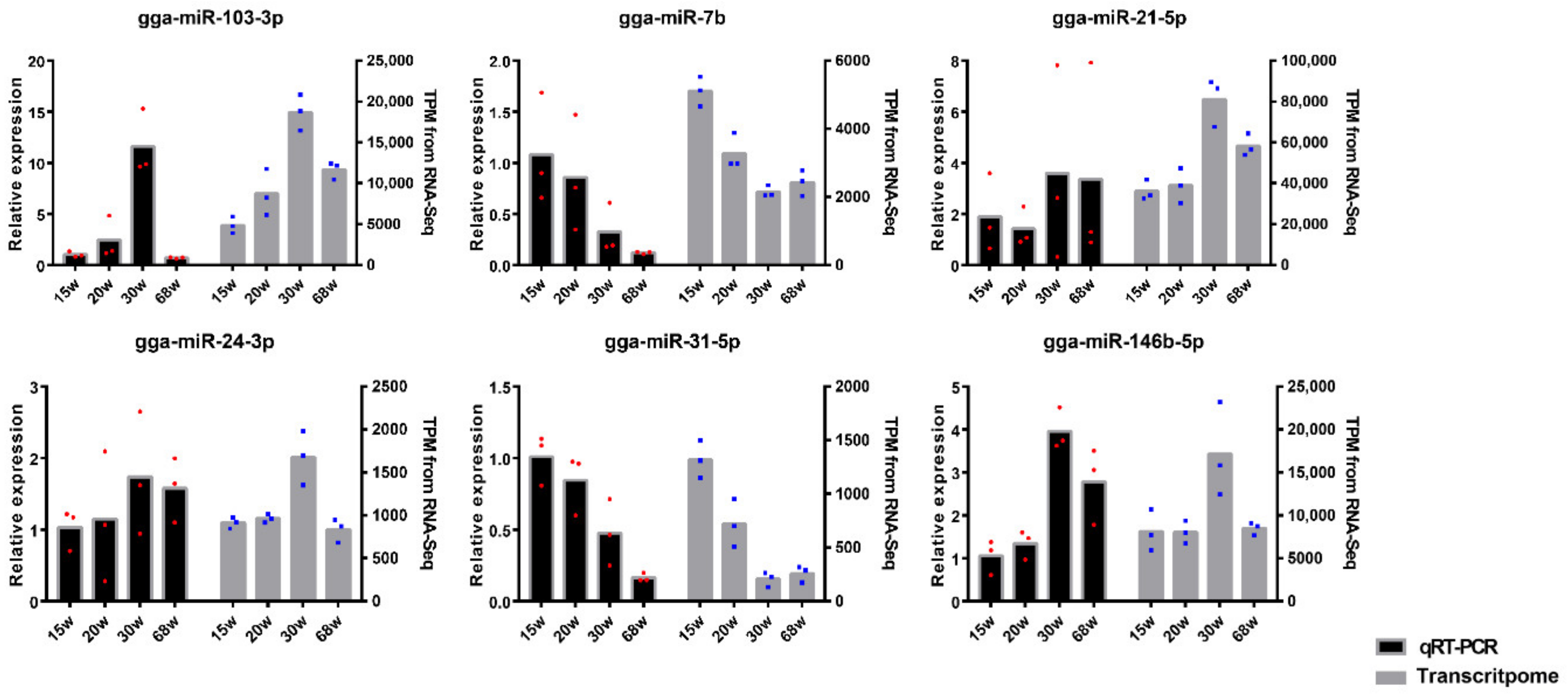
2.7. Validation of miRNA Expression
3. Results
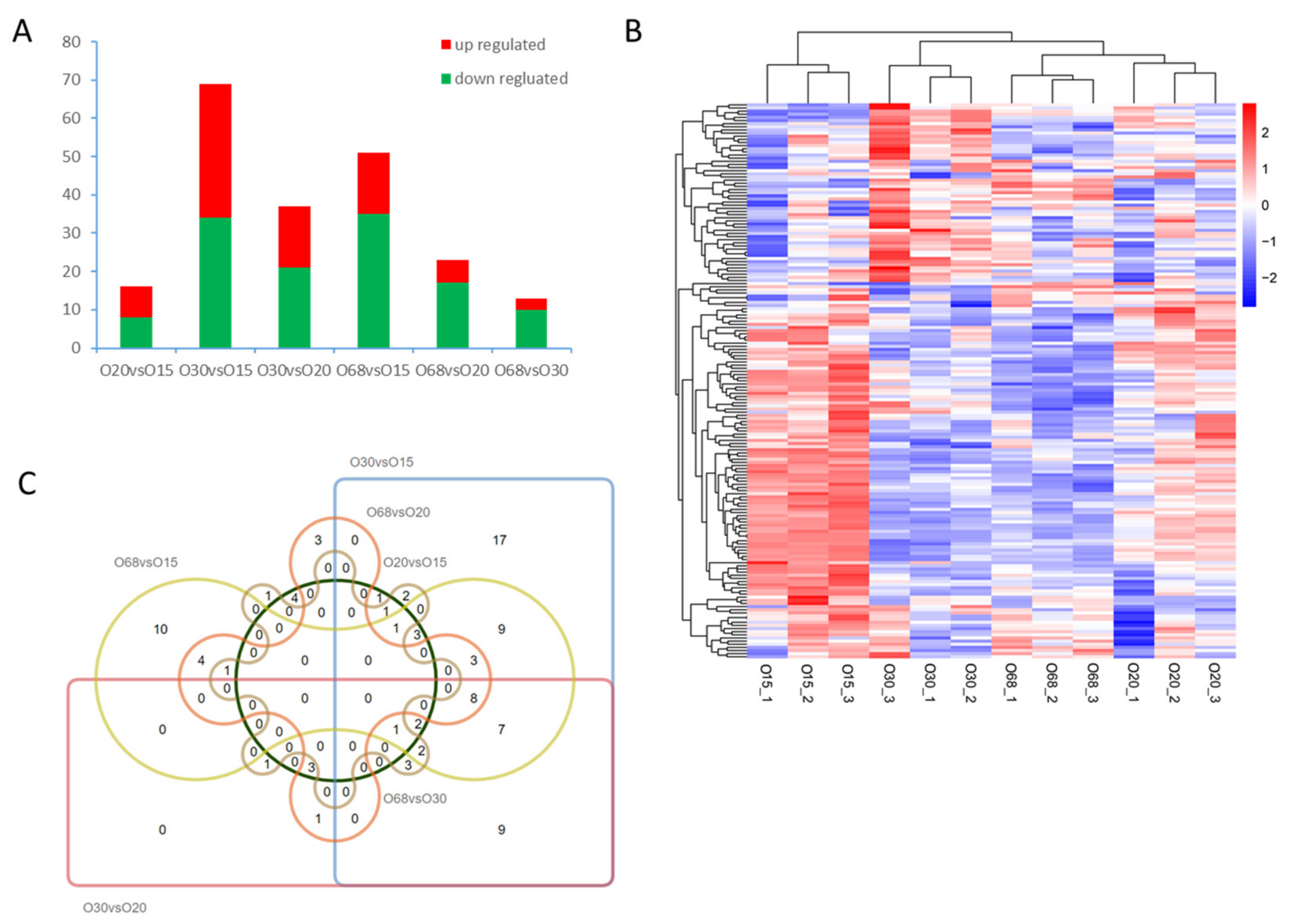
3.1. Sequencing Analysis
3.2. DE miRNA Analysis
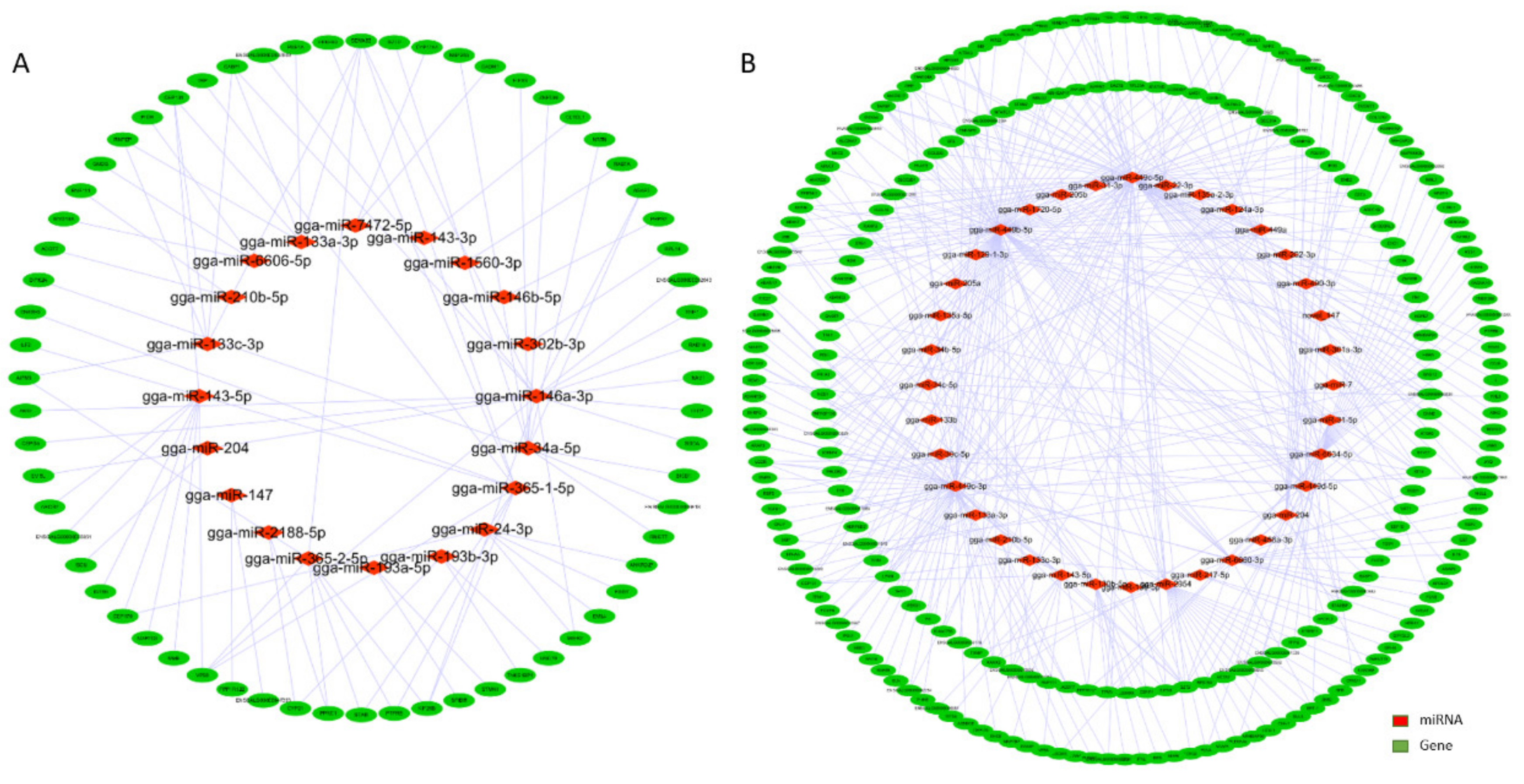
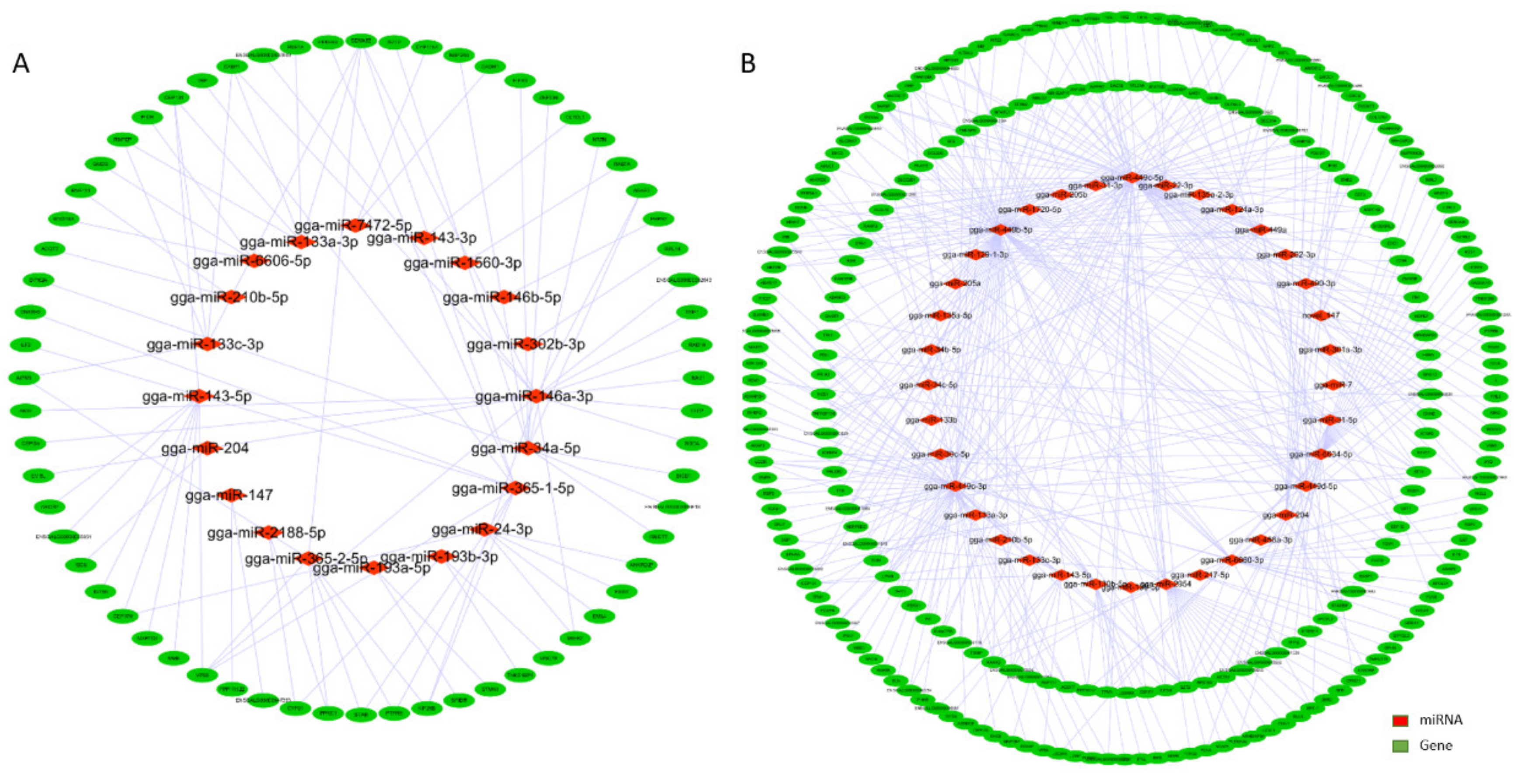
3.3. Interaction Network Analysis of miRNA-mRNA
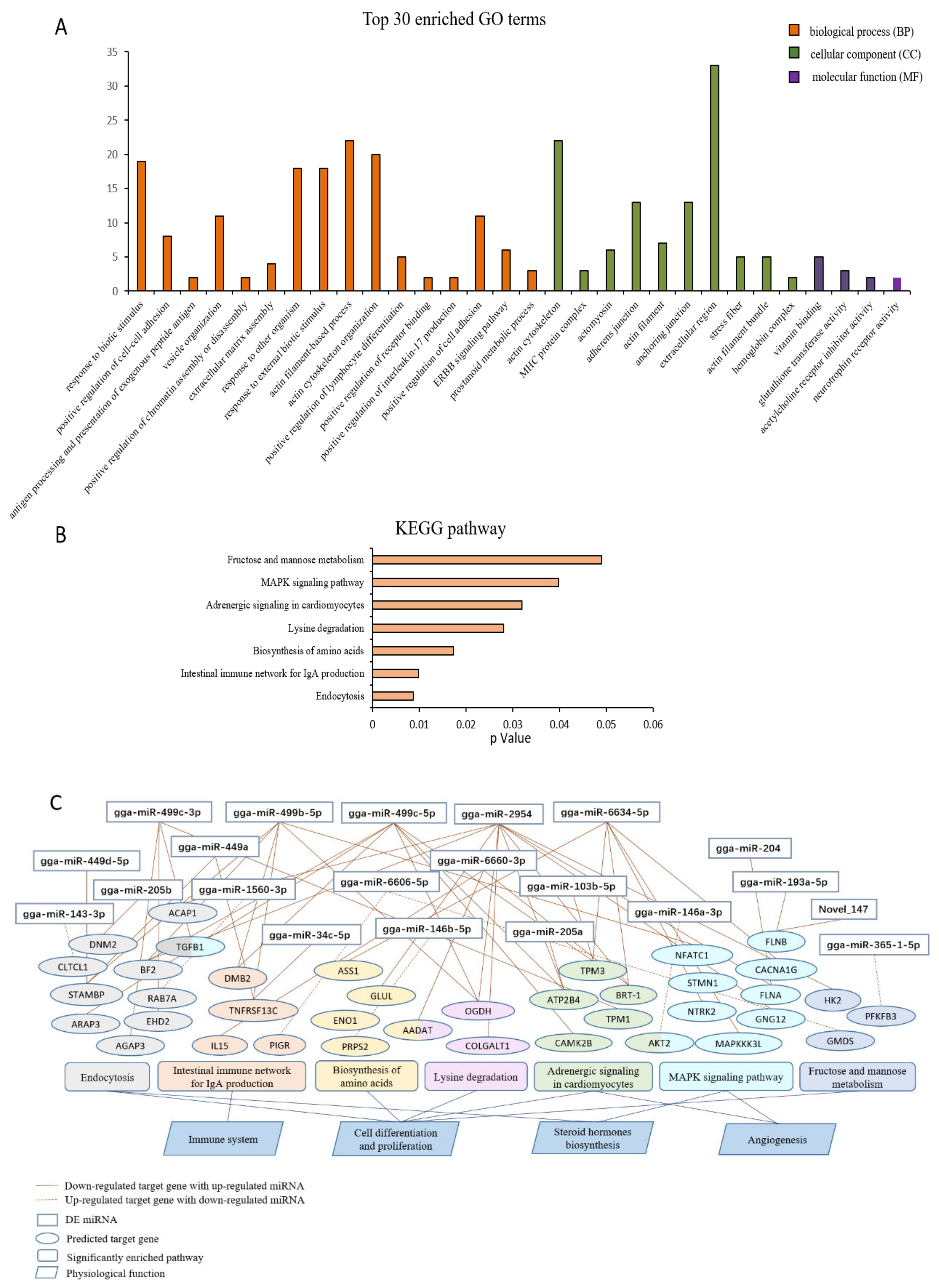
3.4. Functional Enrichment Analysis
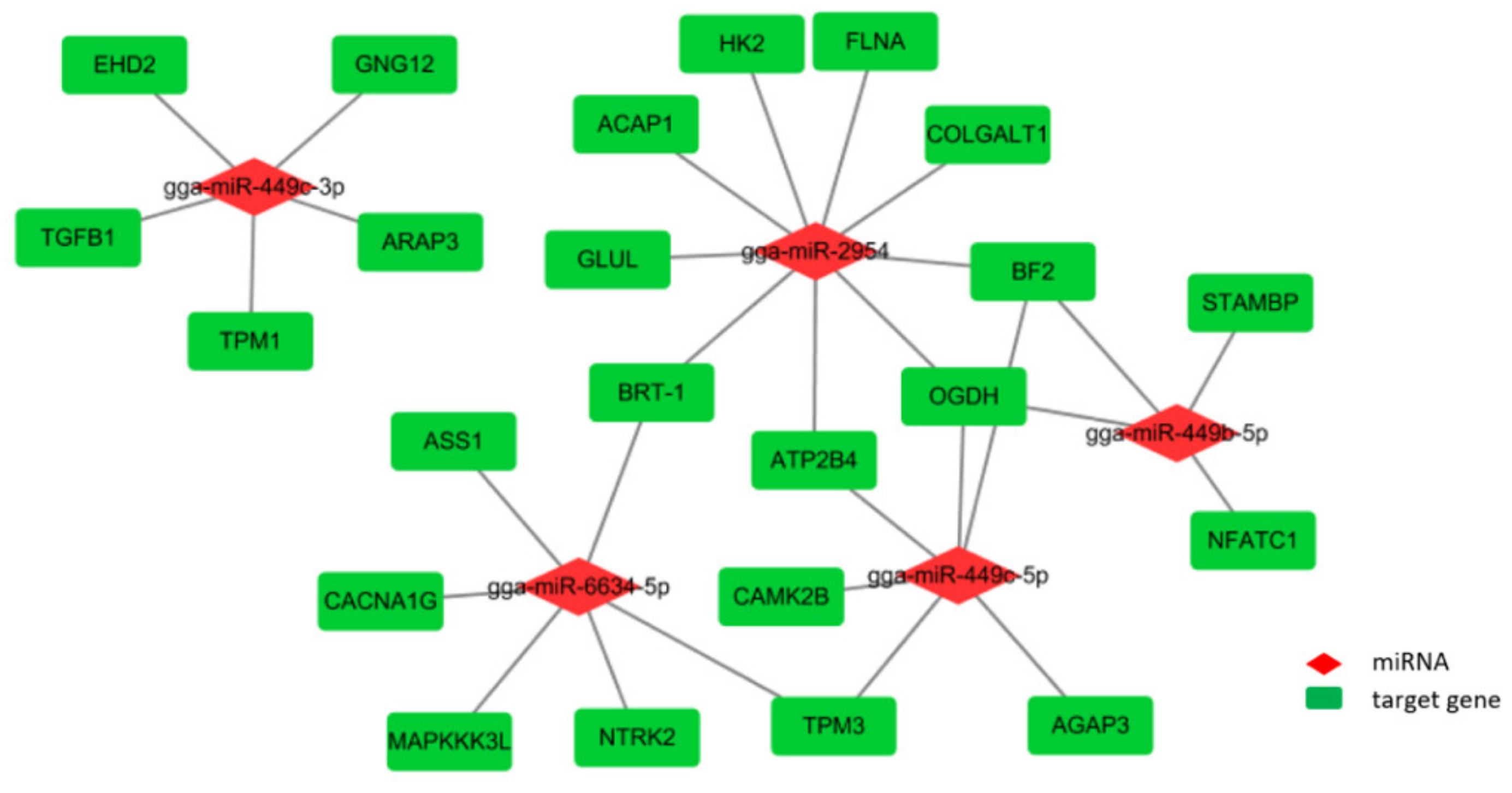
3.5. Integrated Analysis of Key DE miRNAs
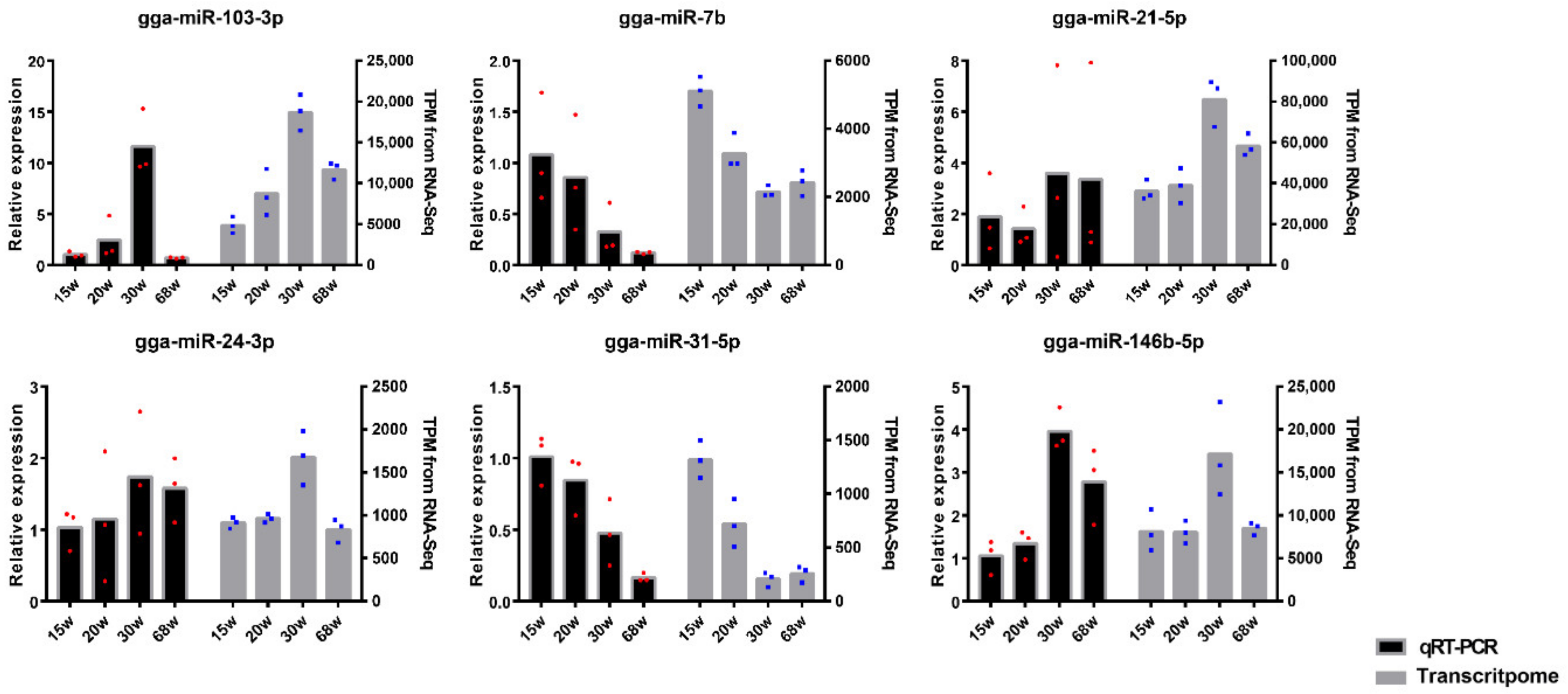
3.6. Validation of miRNA Profiles by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zakaria, A.H.; Miyaki, T.; Imai, K. The Effect of Aging on the Ovarian Follicular Growth in Laying Hens. Poult. Sci. 1983, 62, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Barua, A.; Yoshimura, Y.; Tamura, T. Effects of ageing and oestrogen on the localization of immunoglobulin-containing cells in the chicken ovary. J. Reprod. Fertil. 1998, 114, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gong, Y. Transcription of CYP19A1 is directly regulated by SF-1 in the theca cells of ovary follicles in chicken. Gen. Comp. Endocrinol. 2017, 247, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Onagbesan, O.M.; Vleugels, B.; Buys, N.; Bruggeman, V.; Safi, M.; Decuypere, E. Insulin-like growth factors in the regulation of avian ovarian functions. Domest. Anim. Endocrinol. 1999, 17, 299–313. [Google Scholar] [CrossRef]
- Onagbesan, O.; Bruggeman, V.; Decuypere, E. Intra-ovarian growth factors regulating ovarian function in avian species: A review. Anim. Reprod. Sci. 2009, 111, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Bushati, N.; Cohen, S.M. microRNA Functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, S.W.; Liu, W.; Pate, J.L. MicroRNA in ovarian function. Cell Tissue Res. 2015, 363, 7–18. [Google Scholar] [CrossRef]
- Sen, A.; Prizant, H.; Light, A.; Biswas, A.; Hayes, E.; Lee, H.-J.; Barad, D.; Gleicher, N.; Hammes, S.R. Androgens regulate ovarian follicular development by increasing follicle stimulating hormone receptor and microRNA-125b expression. Proc. Natl. Acad. Sci. USA 2014, 111, 3008–3013. [Google Scholar] [CrossRef] [Green Version]
- Gebremedhn, S.; Salilew-Wondim, D.; Ahmad, I.; Sahadevan, S.; Hossain, M.; Hoelker, M.; Rings, F.; Neuhoff, C.; Tholen, E.; Looft, C.; et al. MicroRNA Expression Profile in Bovine Granulosa Cells of Preovulatory Dominant and Subordinate Follicles during the Late Follicular Phase of the Estrous Cycle. PLoS ONE 2015, 10, e0125912. [Google Scholar] [CrossRef]
- Yerushalmi, G.M.; Salmon-Divon, M.; Ophir, L.; Yung, Y.; Baum, M.; Coticchio, G.; Fadini, R.; Mignini-Renzini, M.; Canto, M.D.; Machtinger, R.; et al. Characterization of the miRNA regulators of the human ovulatory cascade. Sci. Rep. 2018, 8, 15605. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Y.-X.; Liu, H.; Pan, Z. MicroRNAs in ovarian follicular atresia and granulosa cell apoptosis. Reprod. Biol. Endocrinol. (RBE) 2019, 17, 9. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Q.; Qin, R.; Zhang, K.; Li, H. MicroRNA-449a reduces cell survival and enhances cisplatin-induced cytotoxicity via downregulation of NOTCH1 in ovarian cancer cells. Tumor Biol. 2014, 35, 12369–12378. [Google Scholar] [CrossRef]
- Worku, T.; Rehman, Z.U.; Talpur, H.S.; Bhattarai, D.; Ullah, F.; Malobi, N.; Kebede, T.; Yang, L. MicroRNAs: New Insight in Modulating Follicular Atresia: A Review. Int. J. Mol. Sci. 2017, 18, 333. [Google Scholar] [CrossRef] [PubMed]
- Donadeu, F.X.; Sontakke, S.D.; Ioannidis, J. MicroRNA indicators of follicular steroidogenesis. Reprod. Fertil. Dev. 2017, 29, 906. [Google Scholar] [CrossRef]
- Yu, C.; Li, M.; Wang, Y.; Liu, Y.; Yan, C.; Pan, J.; Liu, J.; Cui, S. miR-375 mediates CRH signaling pathway in inhibiting E2 synthesis in porcine ovary. Reproduction 2017, 153, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Yang, C.; Wu, H.; Chen, Q.; Huang, L.; Li, X.; Tang, H.; Jiang, Y. miR-26a-5p Regulates TNRC6A Expression and Facilitates Theca Cell Proliferation in Chicken Ovarian Follicles. DNA Cell Biol. 2017, 36, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, L.; Sun, Y.; Xing, J.; Jiang, Y.; Kang, L. Single nucleotide polymorphism rs737028527 (G>A) affect miR-1b-3p biogenesis and effects on chicken egg-laying traits. Anim. Reprod. Sci. 2020, 218, 106476. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated Profiling of MicroRNAs and mRNAs: MicroRNAs Located on Xq27.3 Associate with Clear Cell Renal Cell Carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [Green Version]
- Robinson, F.E.; Renema, R.A.; Oosterhoff, H.H.; Zuidhof, M.; Wilson, J.L. Carcass Traits, Ovarian Morphology and Egg Laying Characteristics in Early Versus Late Maturing Strains of Commercial Egg-Type Hens. Poult. Sci. 2001, 80, 37–46. [Google Scholar] [CrossRef]
- Johnson, A.L.; Woods, D.C. Dynamics of avian ovarian follicle development: Cellular mechanisms of granulosa cell differentiation. Gen. Comp. Endocrinol. 2009, 163, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Waddington, D.; Perry, M.M.; Gilbert, A.B.; Hardie, M.A. Follicular growth and atresia in the ovaries of hens (Gallus domesticus) with diminished egg production rates. J. Reprod. Fertil. 1985, 74, 399–405. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-Y.; Pan, S.-S. MiR-202-5p suppressed cell proliferation, migration and invasion in ovarian cancer via regulating HOXB2. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2256–2263. [Google Scholar] [PubMed]
- Zhang, Z.; Chen, C.-Z.; Xu, M.-Q.; Zhang, L.-Q.; Liu, J.-B.; Gao, Y.; Jiang, H.; Yuan, B.; Zhang, J.-B. MiR-31 and miR-143 affect steroid hormone synthesis and inhibit cell apoptosis in bovine granulosa cells through FSHR. Theriogenology 2019, 123, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lu, S.; Hu, Y.; Guo, L.; Wu, X.; Liu, X.; Sun, Y. MicroRNA-135a Regulates VEGFC Expression and Promotes Luteinized Granulosa Cell Apoptosis in Polycystic Ovary Syndrome. Reprod. Sci. 2020, 27, 1436–1442. [Google Scholar] [CrossRef]
- Yuan, J.-M.; Shi, X.-J.; Sun, P.; Liu, J.-X.; Wang, W.; Li, M.; Ling, F.-Y. Downregulation of cell cycle-related proteins in ovarian cancer line and cell cycle arrest induced by microRNA. Int. J. Clin. Exp. Med. 2015, 8, 18476–18481. [Google Scholar]
- Luo, G.; Hafner, M.; Shi, Z.; Brown, M.; Feng, G.-H.; Tuschl, T.; Wang, X.-J.; Li, X. Genome-wide annotation and analysis of zebra finch microRNA repertoire reveal sex-biased expression. BMC Genom. 2012, 13, 727. [Google Scholar] [CrossRef] [Green Version]
- Warnefors, M.; Mössinger, K.; Halbert, J.; Studer, T.; VandeBerg, J.L.; Lindgren, I.; Fallahshahroudi, A.; Jensen, P.; Kaessmann, H. Sex-biased microRNA expression in mammals and birds reveals underlying regulatory mechanisms and a role in dosage compensation. Genome Res. 2017, 27, 1961–1973. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hu, S.; Wang, Y.; Deng, Y.; Yang, S.; Hu, J.; Li, L.; Wang, J. mRNA and miRNA Transcriptome Profiling of Granulosa and Theca Layers from Geese Ovarian Follicles Reveals the Crucial Pathways and Interaction Networks for Regulation of Follicle Selection. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Kang, L.; Wei, Q.; Cui, X.; Wang, S.; Chen, Y.; Jiang, Y. Expression and Regulation of MMP1, MMP3, and MMP9 in the Chicken Ovary in Response to Gonadotropins, Sex Hormones, and TGFB11. Biol. Reprod. 2014, 90, 57. [Google Scholar] [CrossRef] [Green Version]
- Rosairo, D.; Kuyznierewicz, I.; Findlay, J.; Drummond, A. Transforming growth factor-beta: Its role in ovarian follicle development. Reproduction 2008, 136, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tang, C.; Yang, C.; Zheng, Q.; Hou, Y. Tropomyosin-1 Functions as a Tumor Suppressor with Respect to Cell Proliferation, Angiogenesis and Metastasis in Renal Cell Carcinoma. J. Cancer 2019, 10, 2220–2228. [Google Scholar] [CrossRef]
- Lebedeva, I.Y.; A Lebedev, V.; Grossmann, R.; Parvizi, N. Age-dependent role of steroids in the regulation of growth of the hen follicular wall. Reprod. Biol. Endocrinol. (RBE) 2010, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Lee, J.; Johnson, A.L. Vascular endothelial growth factor and angiopoietins during hen ovarian follicle development. Gen. Comp. Endocrinol. 2016, 232, 25–31. [Google Scholar] [CrossRef]
- Lizé, M.; Klimke, A.; Dobbelstein, M. MicroRNA-449 in cell fate determination. Cell Cycle 2011, 10, 2874–2882. [Google Scholar] [CrossRef] [Green Version]
- Kheir, T.B.; Futoma-Kazmierczak, E.; Skanderup, A.J.; Krogh, A.; Bardram, L.; Hother, C.; Grønbæk, K.; Federspiel, B.; Lund, A.H.; Friis-Hansen, L. miR-449 inhibits cell proliferation and is down-regulated in gastric cancer. Mol. Cancer 2011, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Sandbothe, M.; Buurman, R.; Reich, N.; Greiwe, L.; Vajen, B.; Gürlevik, E.; Schäffer, V.; Eilers, M.; Kühnel, F.; Vaquero, A.; et al. The microRNA-449 family inhibits TGF-β-mediated liver cancer cell migration by targeting SOX4. J. Hepatol. 2017, 66, 1012–1021. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads | Clean Reads | Clean Ratio (%) | Mapped Reads | Mapped Ratio (%) | Q20 (%) | Frequency Percent of 21–23 nt (%) |
|---|---|---|---|---|---|---|---|
| O15_1 | 12,668,758 | 12,485,845 | 98.56 | 9,568,340 | 94.1 | 99.81 | 79.25 |
| O15_2 | 17,026,099 | 16,850,839 | 98.97 | 11,052,700 | 96.31 | 99.76 | 80.54 |
| O15_3 | 11,521,207 | 11,369,282 | 98.68 | 11,614,036 | 95.84 | 99.69 | 79.78 |
| O20_1 | 13,211,633 | 13,123,238 | 99.33 | 10,360,873 | 94.65 | 99.69 | 77.09 |
| O20_2 | 13,225,084 | 13,073,265 | 98.85 | 10,436,434 | 95.82 | 99.73 | 86.56 |
| O20_3 | 11,191,768 | 11,061,677 | 98.84 | 10,414,140 | 96.03 | 99.7 | 85.07 |
| O30_1 | 12,553,362 | 12,421,066 | 98.95 | 10,578,899 | 95.37 | 99.74 | 83.34 |
| O30_2 | 11,766,007 | 11,652,830 | 99.04 | 11,540,385 | 95.37 | 99.81 | 87.53 |
| O30_3 | 11,300,865 | 11,136,431 | 98.54 | 12,259,060 | 96.56 | 99.7 | 87.78 |
| O68_1 | 16,894,428 | 16,730,280 | 99.03 | 12,135,277 | 96.15 | 99.67 | 85.81 |
| O68_2 | 11,172,817 | 11,062,162 | 99.01 | 15,619,129 | 95.14 | 99.82 | 87.39 |
| O68_3 | 10,345,214 | 10,290,138 | 99.47 | 15,541,471 | 94.27 | 99.85 | 87.72 |
| Average | 12,739,770 | 12,604,754 | 98.94 | 11,760,062 | 95.47 | 99.76 | 83.99 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Li, C.; Li, Q.; Li, W.-T.; Li, H.; Li, G.-X.; Kang, X.-T.; Liu, X.-J.; Tian, Y.-D. Identification of the Key microRNAs and miRNA-mRNA Interaction Networks during the Ovarian Development of Hens. Animals 2020, 10, 1680. https://doi.org/10.3390/ani10091680
Li J, Li C, Li Q, Li W-T, Li H, Li G-X, Kang X-T, Liu X-J, Tian Y-D. Identification of the Key microRNAs and miRNA-mRNA Interaction Networks during the Ovarian Development of Hens. Animals. 2020; 10(9):1680. https://doi.org/10.3390/ani10091680
Chicago/Turabian StyleLi, Jing, Chong Li, Qi Li, Wen-Ting Li, Hong Li, Guo-Xi Li, Xiang-Tao Kang, Xiao-Jun Liu, and Ya-Dong Tian. 2020. "Identification of the Key microRNAs and miRNA-mRNA Interaction Networks during the Ovarian Development of Hens" Animals 10, no. 9: 1680. https://doi.org/10.3390/ani10091680
APA StyleLi, J., Li, C., Li, Q., Li, W.-T., Li, H., Li, G.-X., Kang, X.-T., Liu, X.-J., & Tian, Y.-D. (2020). Identification of the Key microRNAs and miRNA-mRNA Interaction Networks during the Ovarian Development of Hens. Animals, 10(9), 1680. https://doi.org/10.3390/ani10091680