Transcriptomic and Morphological Analysis of Cells Derived from Porcine Buccal Mucosa—Studies on an In Vitro Model

 , ,
, ,  , ,
, ,  ,
, 


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Buccal Mucosal Cells Isolation and In Vitro Culture
2.3. Microarray Expression Analysis and Statistics
2.4. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) Analysis
2.5. Automated Morphological Analysis of DIC Imaging
2.5.1. Image Processing and Segmentation
2.5.2. Morphological Analysis
2.6. Confocal Microscopic Observations of pCK Expression and Distribution
3. Results
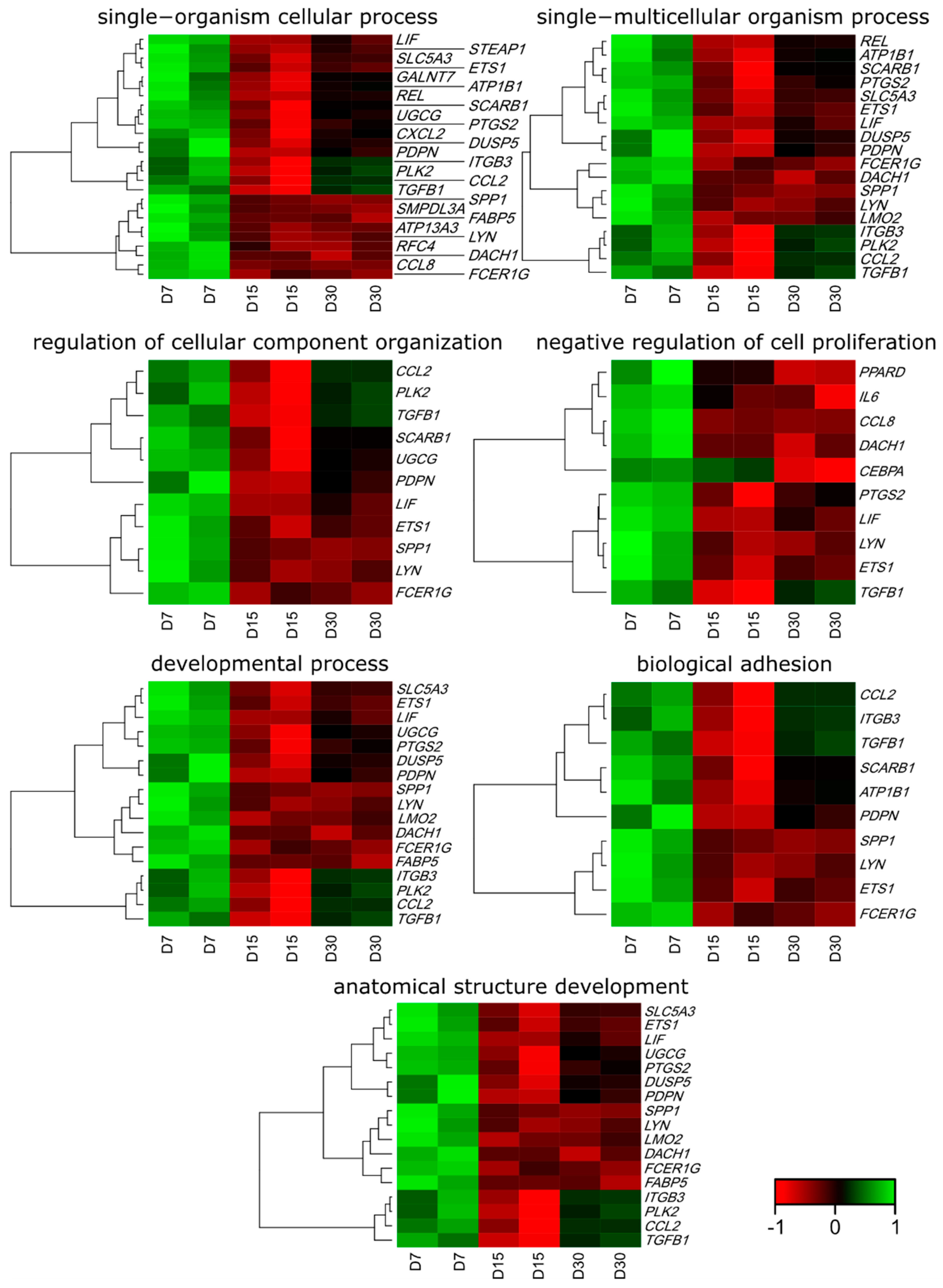
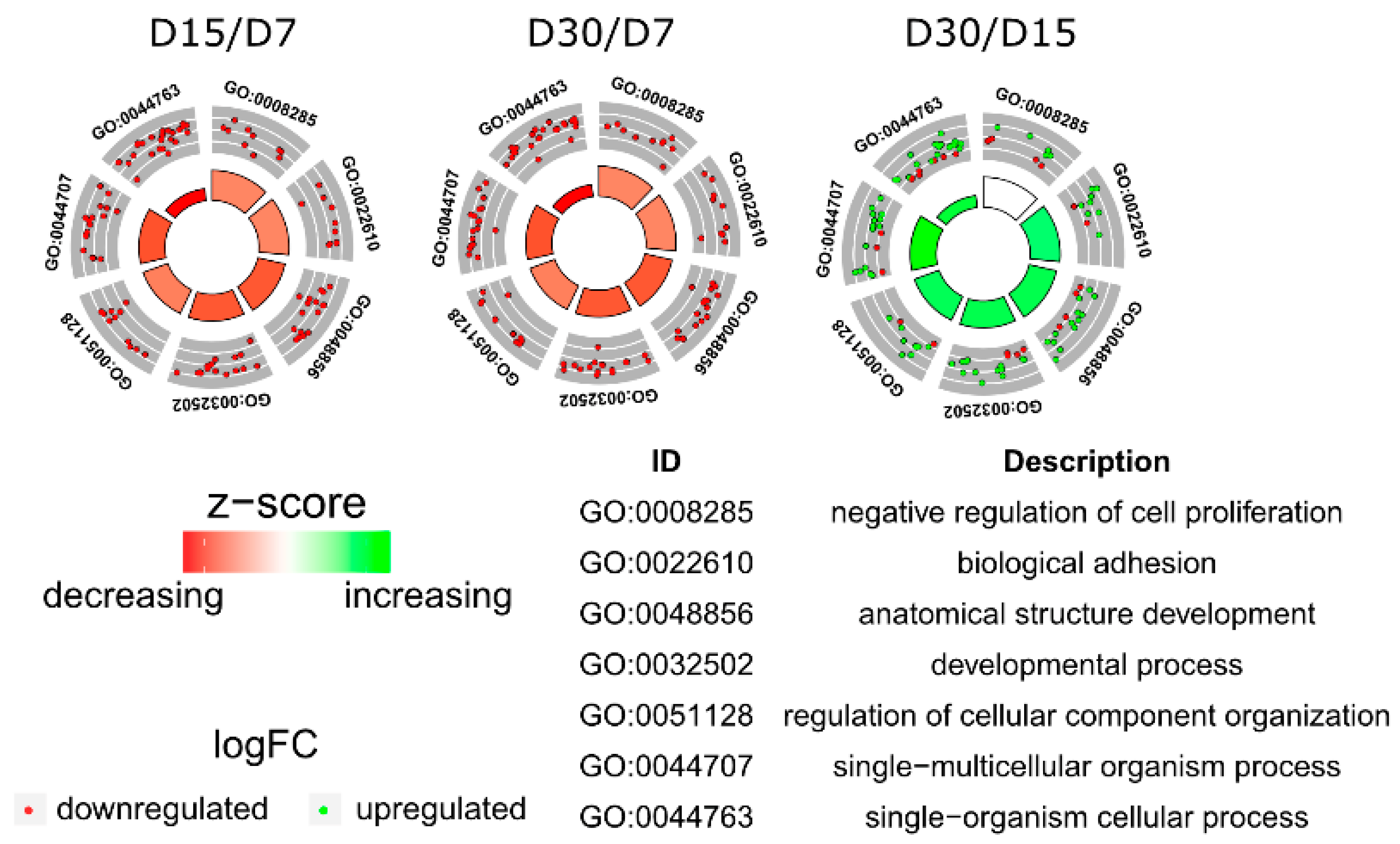
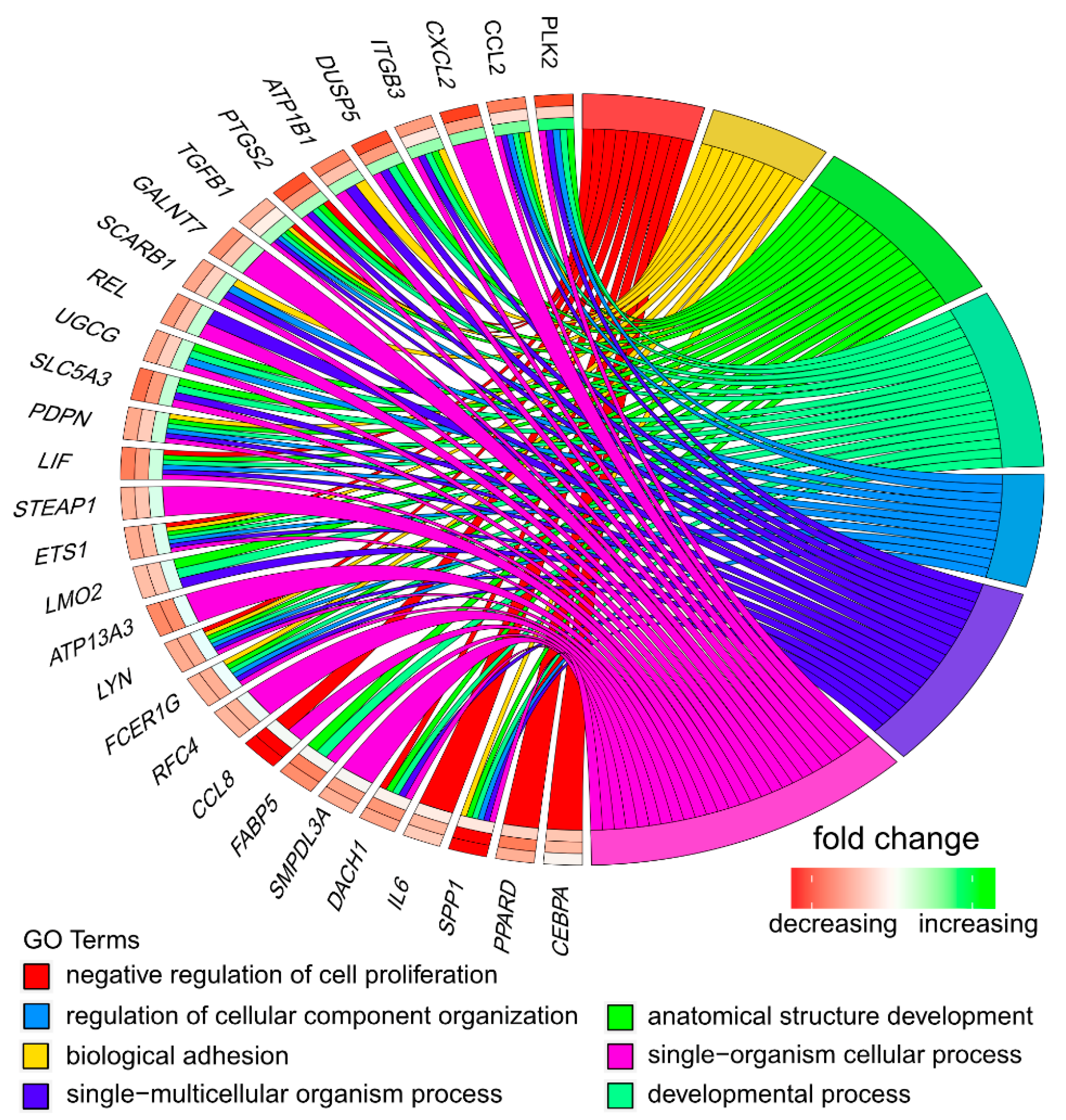
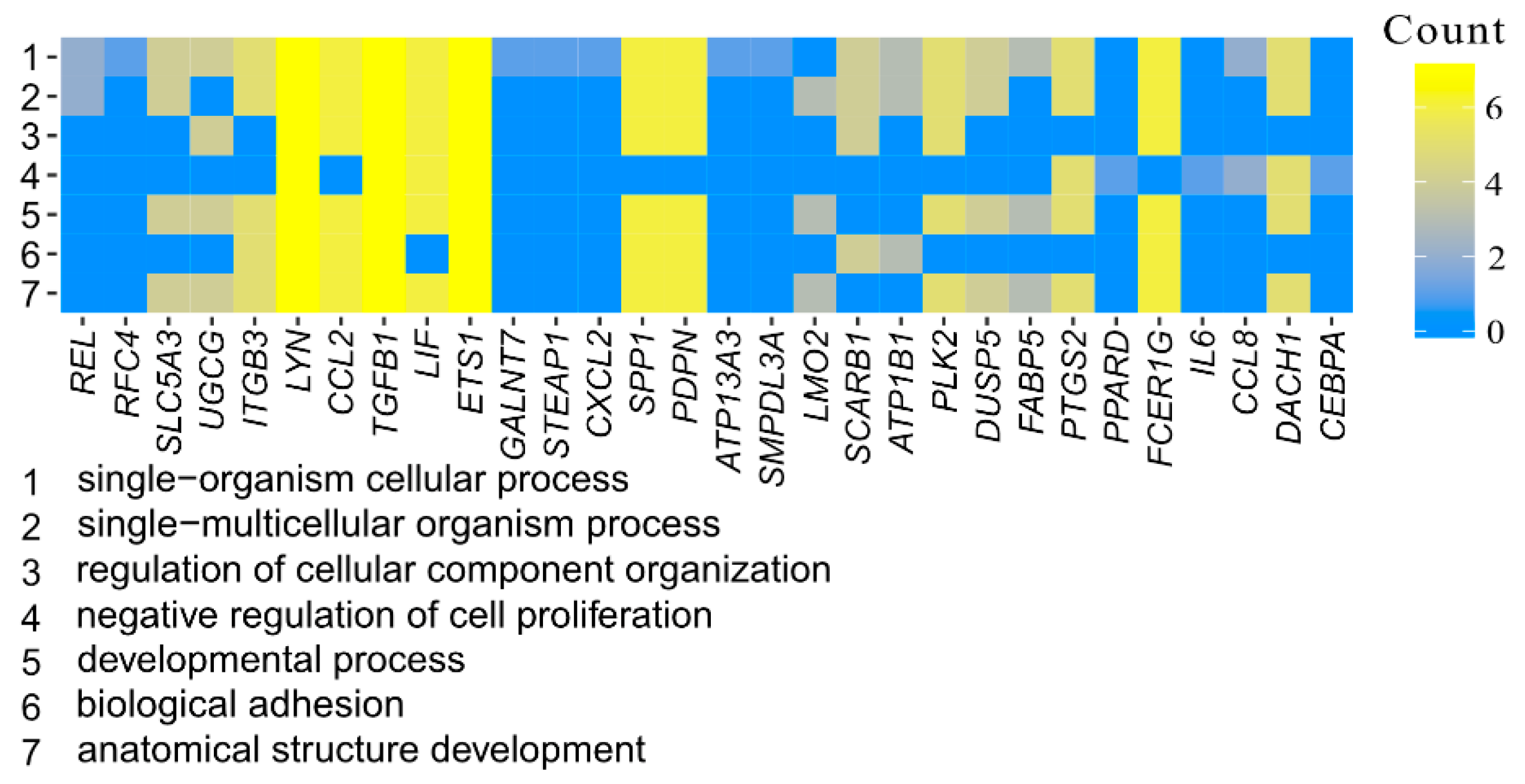
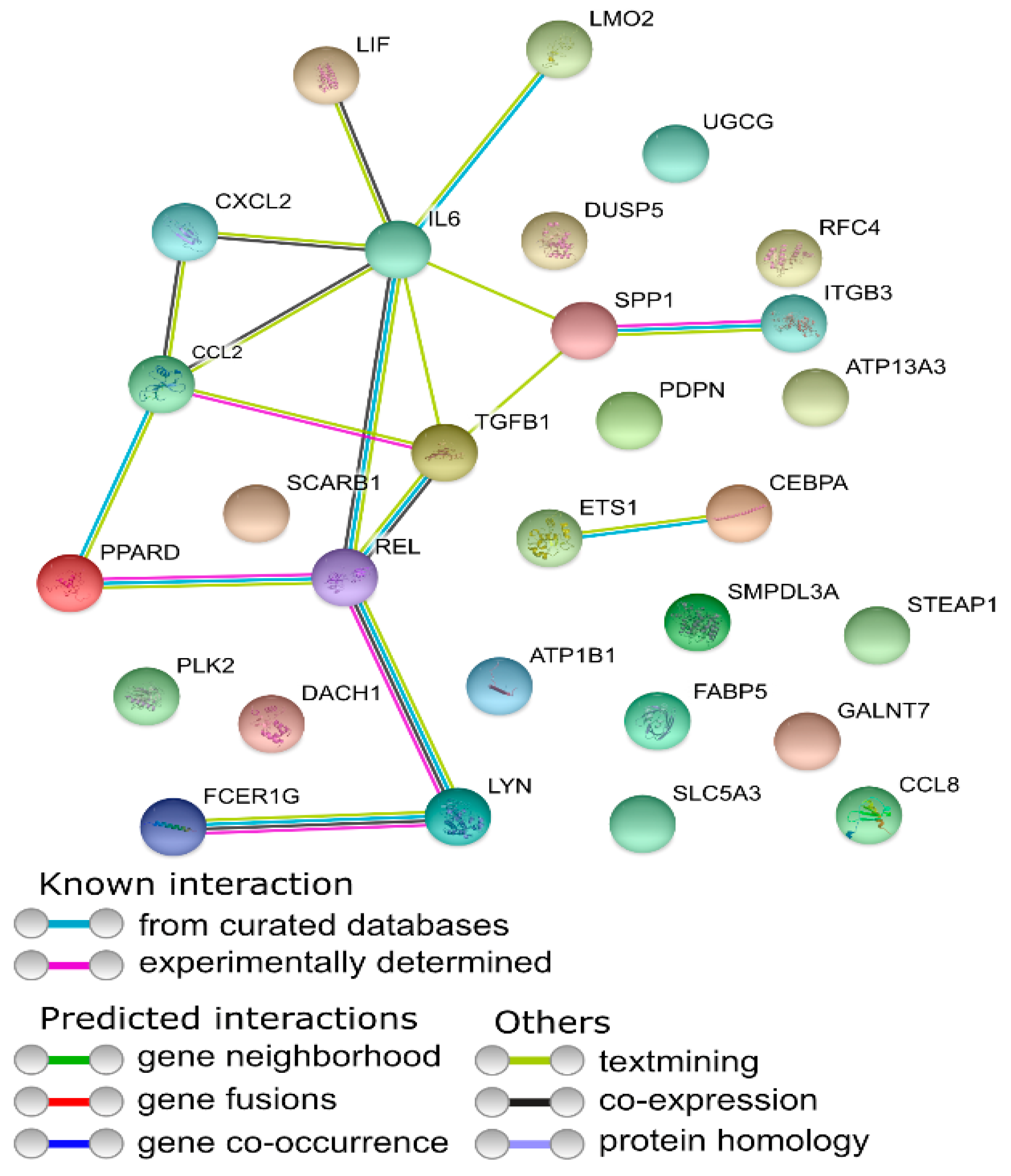
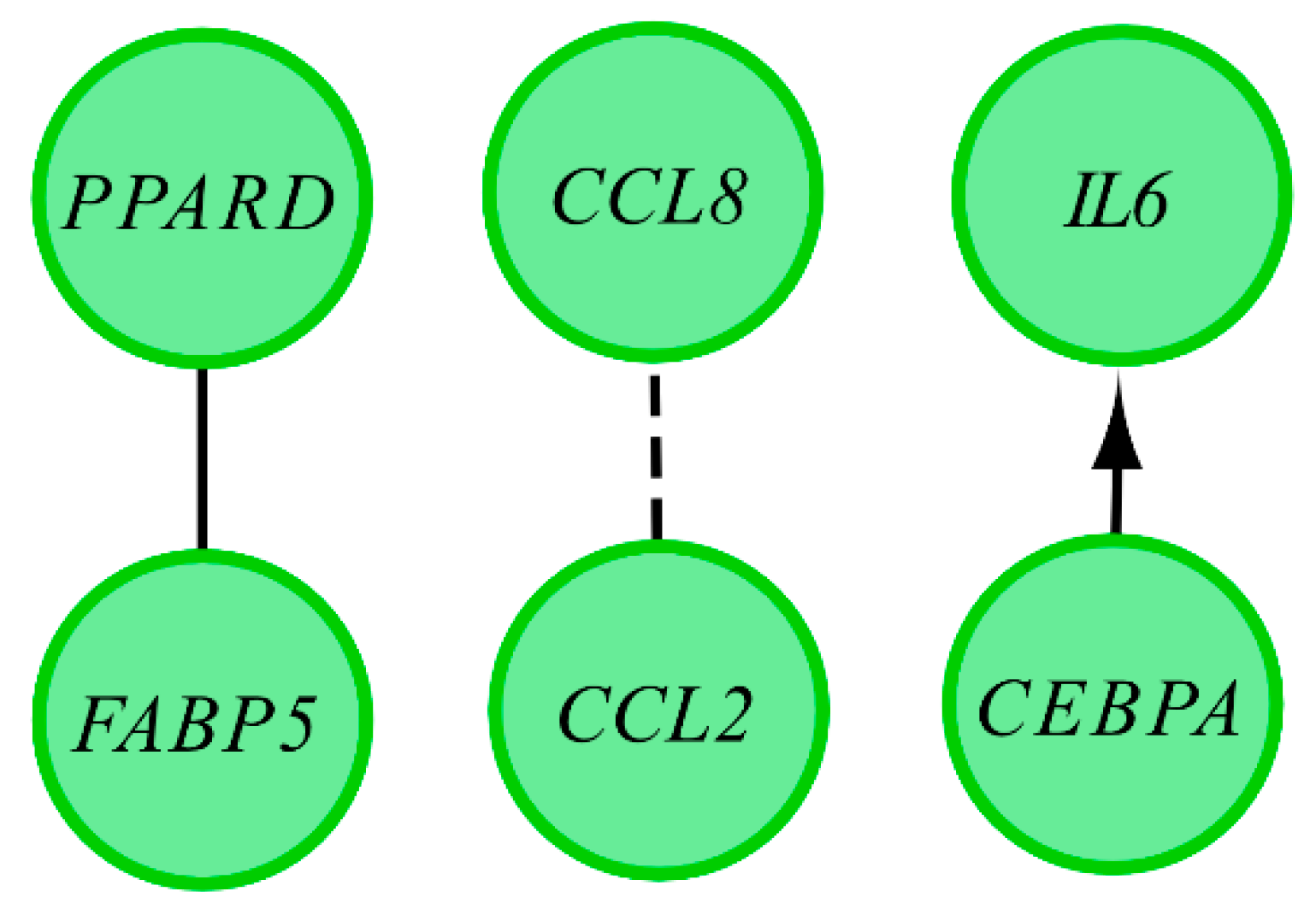
3.1. Microarray Analysis
3.2. RT-qPCR Analysis
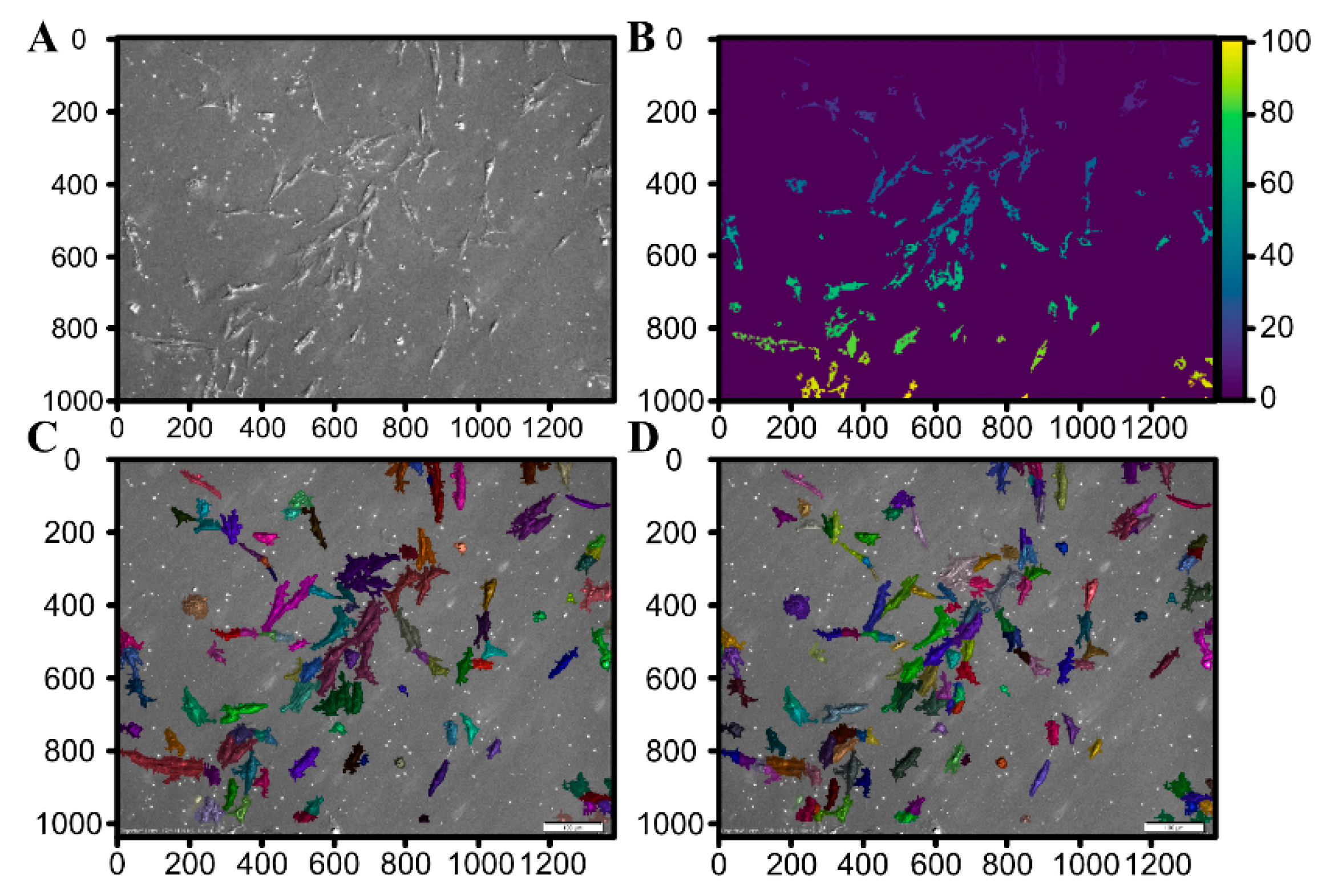
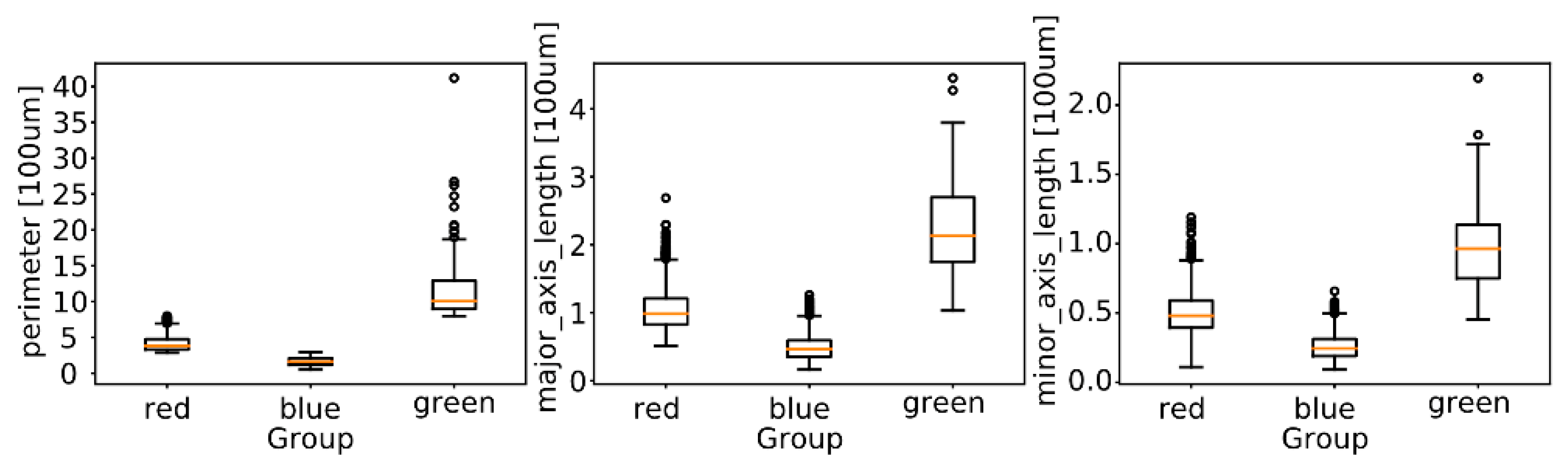
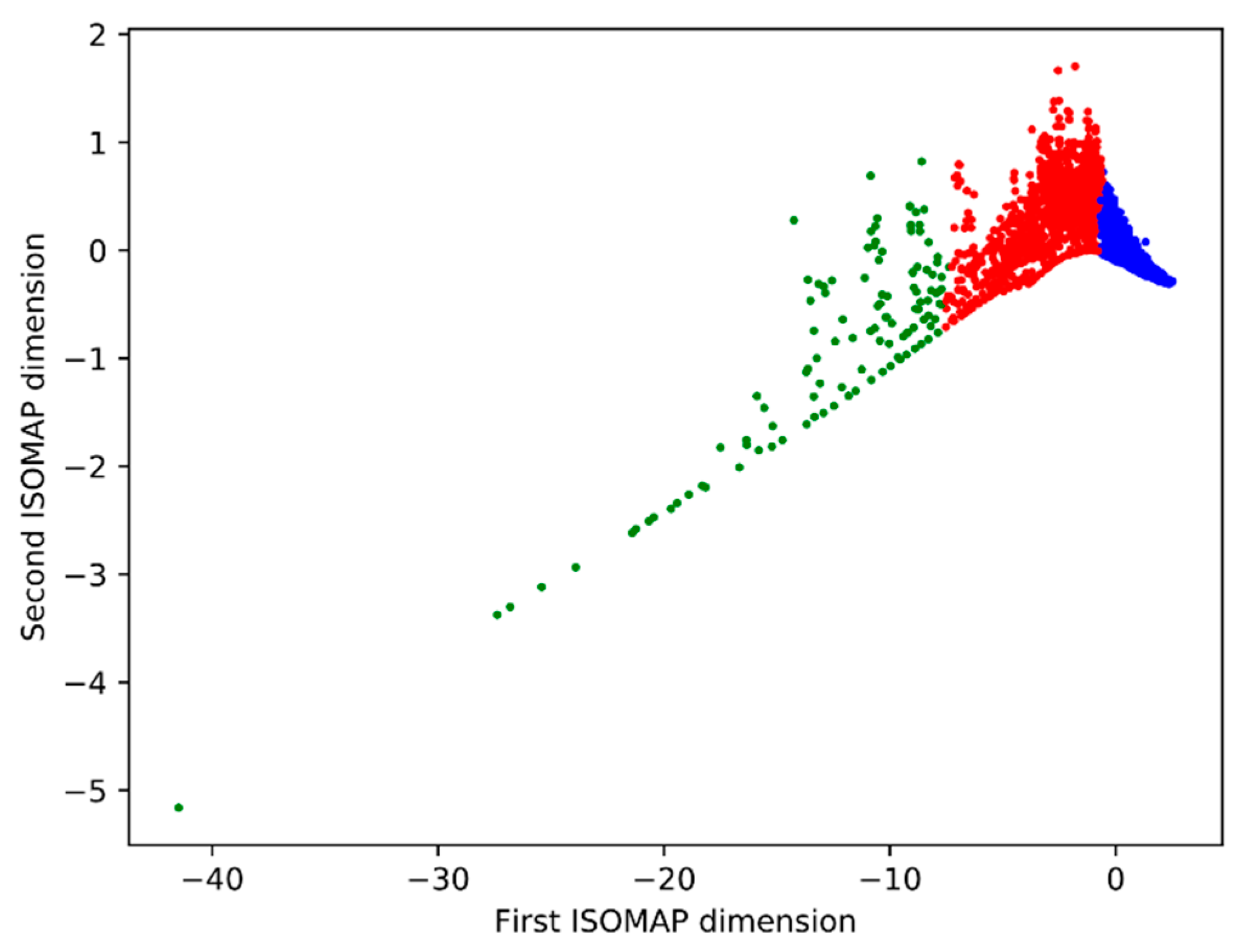
3.3. Morphological Analysis
3.4. Confocal Microscope Observations of pCK Expression and Distribution
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bryja, A.; Dyszkiewicz-Konwińska, M.; Budna, J.; Kranc, W.; Chachuła, A.; Borys, S.; Ciesiółka, S.; Sokalski, J.; Pryliński, M.; Bukowska, D.; et al. The biomedical aspects of oral mucosal epithelial cell culture in mammals. J. Biol. Regul. Homeost. Agents 2017, 31, 81–85. [Google Scholar] [PubMed]
- Swindle, M.M.; Makin, A.; Herron, A.J.; Clubb, F.J.; Frazier, K.S. Swine as models in biomedical research and toxicology testing. Vet. Pathol. 2012, 49, 344–356. [Google Scholar] [CrossRef]
- Hunter, P. Xeno’s paradox. Why pig cells are better for tissue transplants than human cells. EMBO Rep. 2009, 10, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Denner, J. Paving the path toward porcine organs for transplantation. N. Engl. J. Med. 2017, 377, 1891–1893. [Google Scholar] [CrossRef] [PubMed]
- McGregor, C.G.A.; Byrne, G.W. Porcine to human heart transplantation: Is clinical application now appropriate? J. Immunol. Res. 2017, 2017, 2534653. [Google Scholar] [CrossRef] [Green Version]
- Mohiuddin, M.M.; Reichart, B.; Byrne, G.W.; McGregor, C.G.A. Current status of pig heart xenotransplantation. Int. J. Surg. 2015, 23, 234–239. [Google Scholar] [CrossRef]
- Nagashima, H.; Matsunari, H. Growing human organs in pigs-A dream or reality? Theriogenology 2016, 86, 422–426. [Google Scholar] [CrossRef] [Green Version]
- Leonard, D.A.; Mallard, C.; Albritton, A.; Torabi, R.; Mastroianni, M.; Sachs, D.H.; Kurtz, J.M.; Cetrulo, C.L. Skin grafts from genetically modified α-1,3-galactosyltransferase knockout miniature swine: A functional equivalent to allografts. Burns 2017, 43, 1717–1724. [Google Scholar] [CrossRef] [Green Version]
- Albritton, A.; Leonard, D.A.; Leto Barone, A.; Keegan, J.; Mallard, C.; Sachs, D.H.; Kurtz, J.M.; Cetrulo, C.L. Lack of cross-sensitization between alpha-1,3-galactosyltransferase knockout porcine and allogeneic skin grafts permits serial grafting. Transplantation 2014, 97, 1209–1215. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, K.; Dicks, N.; Glanzner, W.G.; Agellon, L.B.; Bordignon, V. Efficacy of the porcine species in biomedical research. Front. Genet. 2015, 6, 293. [Google Scholar] [CrossRef] [Green Version]
- Albl, B.; Haesner, S.; Braun-Reichhart, C.; Streckel, E.; Renner, S.; Seeliger, F.; Wolf, E.; Wanke, R.; Blutke, A. Tissue sampling guides for porcine biomedical models. Toxicol. Pathol. 2016, 44, 414–420. [Google Scholar] [CrossRef]
- Rojewska, M.; Popis, M.; Jankowski, M.; Bukowska, D.; Antosik, P.; Kempisty, B. Stemness specificity of epithelial cells—Application of cell and tissue technology in regenerative medicine. Med. J. Cell Biol. 2018, 6, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, S.; Roffel, S.; Meyer, M.; Gasser, A. Biology of soft tissue repair: Gingival epithelium in wound healing and attachment to the tooth and abutment surface. Eur. Cell. Mater. 2019, 38, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Politis, C.; Schoenaers, J.; Jacobs, R.; Agbaje, J.O. Wound healing problems in the mouth. Front. Physiol. 2016, 7, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues Neves, C.; Buskermolen, J.; Roffel, S.; Waaijman, T.; Thon, M.; Veerman, E.; Gibbs, S. Human saliva stimulates skin and oral wound healing in vitro. J. Tissue Eng. Regen. Med. 2019, 13, 1079–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamier-Gliszczyńska, A.; Brązert, M.; Sujka-Kordowska, P.; Popis, M.; Ożegowska, K.; Stefańska, K.; Kocherova, I.; Celichowski, P.; Kulus, M.; Bukowska, D.; et al. Genes involved in angiogenesis and circulatory system development are differentially expressed in porcine epithelial oviductal cells during long-term primary in vitro culture—A transcriptomic study. Med. J. Cell Biol. 2018, 6, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Bryja, A.; Dyszkiewicz-Konwińska, M.; Jankowski, M.; Celichowski, P.; Stefańska, K.; Chamier-Gliszczyńska, A.; Borowiec, B.; Mehr, K.; Bukowska, D.; Antosik, P.; et al. Cation homeostasis and transport related gene markers are differentially expressed in porcine buccal pouch mucosal cells during long-term cells primary culture in vitro. Med. J. Cell Biol. 2018, 6, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Borys-Wójcik, S.; Kocherova, I.; Celichowski, P.; Popis, M.; Jeseta, M.; Bukowska, D.; Antosik, P.; Nowicki, M.; Kempisty, B. Protein oligomerization is the biochemical process highly up-regulated in porcine oocytes before in vitro maturation (IVM). Med. J. Cell Biol. 2018, 6, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, M.J.; Celichowski, P.; Jankowski, M.; Kranc, W.; Bryja, A.; Borys-Wójcik, S.; Jeseta, M.; Antosik, P.; Bukowska, D.; Bruska, M.; et al. Ontology groups representing angiogenesis and blood vessels development are highly up-regulated during porcine oviductal epithelial cells long-term real-time proliferation—A primary cell culture approach. Med. J. Cell Biol. 2018, 6, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yan, C.; Hu, Y.; Mu, L.; Huang, K.; Li, Q.; Li, X.; Tao, D.; Qin, J. Sphere-forming assay vs. organoid culture: Determining long-term stemness and the chemoresistant capacity of primary colorectal cancer cells. Int. J. Oncol. 2019, 54, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Bono, B.; Ostano, P.; Peritore, M.; Gregnanin, I.; Belgiovine, C.; Liguori, M.; Allavena, P.; Chiorino, G.; Chiodi, I.; Mondello, C. Cells with stemness features are generated from in vitro transformed human fibroblasts. Sci. Rep. 2018, 8, 13838. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Nowak-Imialek, M.; Chen, X.; Chen, D.; Herrmann, D.; Ruan, D.; Chen, A.C.H.; Eckersley-Maslin, M.A.; Ahmad, S.; Lee, Y.L.; et al. Establishment of porcine and human expanded potential stem cells. Nat. Cell Biol. 2019, 21, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughey, J.J.; Lee, T.K.; Covert, M.W. Computational modeling of mammalian signaling networks. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 194–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, R.; Rhee, A.; Wang, C.J.; Nemenman, I.; Levchenko, A. Information transduction capacity of noisy biochemical signaling networks. Science 2011, 334, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, K.; Ishiwata, H.; Sakuragi, Y.; Hayashi, Y.; Fukuda, A.; Hisatake, K. Live-cell imaging of subcellular structures for quantitative evaluation of pluripotent stem cells. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- van Valen, D.A.; Kudo, T.; Lane, K.M.; Macklin, D.N.; Quach, N.T.; DeFelice, M.M.; Maayan, I.; Tanouchi, Y.; Ashley, E.A.; Covert, M.W. Deep learning automates the quantitative analysis of individual cells in live-cell imaging experiments. PLoS Comput. Biol. 2016, 12, e1005177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzwarth, G.; Webb, S.C.; Kubinski, D.J.; Allen, N.S. Improving DIC microscopy with polarization modulation. J. Microsc. 1997, 188, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Holzwarth, G.M.; Hill, D.B.; McLaughlin, E.B. Polarization-modulated differential-interference contrast microscopy with a variable retarder. Appl. Opt. 2000, 39, 6288–6294. [Google Scholar] [CrossRef]
- Chomczyński, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef]
- Walter, W.; Sanchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinforma. Oxf. Engl. 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef] [PubMed]
- Bradski, G.; Kaehler, A. Learning OpenCV, 1st ed.; Software That Sees; O’Reilly: Beijing, China, 2008; ISBN 978-0-596-51613-0. [Google Scholar]
- van der Walt, S.; Schonberger, J.L.; Nunez-Iglesias, J.; Boulogne, F.; Warner, J.D.; Yager, N.; Gouillart, E.; Yu, T. scikit-image: Image processing in Python. PeerJ 2014, 2, e453. [Google Scholar] [CrossRef]
- Beucher, S.; Lantuéjoul, C. Use of watersheds in contour detection. In Proceedings of the Workshop on Image Processing, CCETT/IRISA, Rennes, France, 17–21 September 1979. [Google Scholar]
- Otsu, N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Feineigle, P.A.; Witkin, A.P.; Stonick, V.L. Processing of 3D DIC microscopy images for data visualization. In Proceedings of the 1996 IEEE International Conference on Acoustics, Speech, and Signal Processing Conference Proceedings, Atlanta, GA, USA, 9 May 1996; IEEE Service Center: Piscataway, NJ, USA, 1996; pp. 2160–2163. [Google Scholar]
- Tenenbaum, J.B.; Silva, V.; Langford, J.C. A global geometric framework for nonlinear dimensionality reduction. Science 2000, 290, 2319–2323. [Google Scholar] [CrossRef]
- Soroka, Y.; Ma’or, Z.; Leshem, Y.; Verochovsky, L.; Neuman, R.; Bregegere, F.M.; Milner, Y. Aged keratinocyte phenotyping: Morphology, biochemical markers and effects of Dead Sea minerals. Exp. Gerontol. 2008, 43, 947–957. [Google Scholar] [CrossRef]
- Ravikanth, M.; Soujanya, P.; Manjunath, K.; Saraswathi, T.R.; Ramachandran, C.R. Heterogenecity of fibroblasts. J. Oral Maxillofac. Pathol. JOMFP 2011, 15, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higa, K.; Satake, Y.; Shimazaki, J. The characterization of human oral mucosal fibroblasts and their use as feeder cells in cultivated epithelial sheets. Future Sci. OA 2017, 3, FSO243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Zhang, Y.; Xu, B.; Li, X. Image Analysis and Processing of Skin Cell Injury Based on OpenCV. J. Phys. Conf. Ser. 2019, 1237, 032003. [Google Scholar] [CrossRef]
- Meimban, R.J.; Fernando, A.R.; Monsura, A.; Ranada, J.; Apduhan, J.C. Blood Cells Counting using Python OpenCV. In Proceedings of the 2018 14th IEEE International Conference on Signal Processing (ICSP), Beijing, China, 12–16 August 2018; pp. 50–53. [Google Scholar]
- Herusutopo, A.; Bisono, R.W.; Meliala, J.I. Application of malaria detection of drawing blood cells using microscopic openCV. CommIT Commun. Inf. Technol. J. 2011, 5, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Jaccard, N.; Griffin, L.D.; Keser, A.; Macown, R.J.; Super, A.; Veraitch, F.S.; Szita, N. Automated method for the rapid and precise estimation of adherent cell culture characteristics from phase contrast microscopy images. Biotechnol. Bioeng. 2014, 111, 504–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Iakova, P.; Wilde, M.; Welm, A.; Goode, T.; Roesler, W.J.; Timchenko, N.A. C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol. Cell 2001, 8, 817–828. [Google Scholar] [CrossRef]
- Lopez, R.G.; Garcia-Silva, S.; Moore, S.J.; Bereshchenko, O.; Martinez-Cruz, A.B.; Ermakova, O.; Kurz, E.; Paramio, J.M.; Nerlov, C. C/EBPalpha and beta couple interfollicular keratinocyte proliferation arrest to commitment and terminal differentiation. Nat. Cell Biol. 2009, 11, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Badache, A.; Hynes, N.E. Interleukin 6 inhibits proliferation and, in cooperation with an epidermal growth factor receptor autocrine loop, increases migration of t47d breast cancer cells. Cancer Res. 2001, 61, 383–391. [Google Scholar]
- Hernandez-Quintero, M.; Kuri-Harcuch, W.; Gonzalez Robles, A.; Castro-Munozledo, F. Interleukin-6 promotes human epidermal keratinocyte proliferation and keratin cytoskeleton reorganization in culture. Cell Tissue Res. 2006, 325, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Shin, D.; Roh, J.-L. Use of a pre-vascularised oral mucosal cell sheet for promoting cutaneous burn wound healing. Theranostics 2018, 8, 5703–5712. [Google Scholar] [CrossRef]
- Zaveri, N.T.; Sato, B.G.; Jiang, F.; Calaoagan, J.; Laderoute, K.R.; Murphy, B.J. A novel peroxisome proliferator-activated receptor delta antagonist, SR13904, has anti-proliferative activity in human cancer cells. Cancer Biol. Ther. 2009, 8, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Schmuth, M.; Haqq, C.M.; Cairns, W.J.; Holder, J.C.; Dorsam, S.; Chang, S.; Lau, P.; Fowler, A.J.; Chuang, G.; Moser, A.H.; et al. Peroxisome proliferator-activated receptor (PPAR)-beta/delta stimulates differentiation and lipid accumulation in keratinocytes. J. Investig. Dermatol. 2004, 122, 971–983. [Google Scholar] [CrossRef] [Green Version]
- Mao-Qiang, M.; Fowler, A.J.; Schmuth, M.; Lau, P.; Chang, S.; Brown, B.E.; Moser, A.H.; Michalik, L.; Desvergne, B.; Wahli, W.; et al. Peroxisome-proliferator-activated receptor (PPAR)-gamma activation stimulates keratinocyte differentiation. J. Investig. Dermatol. 2004, 123, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Graf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [Green Version]
- Bryja, A.; Dyszkiewicz-Konwińska, M.; Jankowski, M.; Celichowski, P.; Stefańska, K.; Chamier-Gliszczyńska, A.; Popis, M.; Mehr, K.; Bukowska, D.; Antosik, P.; et al. Ion homeostasis and transport are regulated by genes differentially expressed in porcine buccal pouch mucosal cells during long-term culture in vitro—A microarray approach. Med. J. Cell Biol. 2018, 6, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Leydon, C.; Imaizumi, M.; Bartlett, R.S.; Wang, S.F.; Thibeault, S.L. Epithelial cells are active participants in vocal fold wound healing: An in vivo animal model of injury. PLoS ONE 2014, 9, e115389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, P.; Chin, S.S.; Wang, D.; Liu, S.; Sinha, S.; Garrett-Sinha, L.A. Ets1 blocks terminal differentiation of keratinocytes and induces expression of matrix metalloproteases and innate immune mediators. J. Cell Sci. 2010, 123, 3566–3575. [Google Scholar] [CrossRef] [Green Version]
- Joseloff, E.; Cataisson, C.; Aamodt, H.; Ocheni, H.; Blumberg, P.; Kraker, A.J.; Yuspa, S.H. Src family kinases phosphorylate protein kinase C delta on tyrosine residues and modify the neoplastic phenotype of skin keratinocytes. J. Biol. Chem. 2002, 277, 12318–12323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, S.; Gamas, P.; Belhacène, N.; Grosso, S.; Pradelli, L.A.; Colosetti, P.; Johansen, C.; Iversen, L.; Deckert, M.; Luciano, F.; et al. The caspase-cleaved form of LYN mediates a psoriasis-like inflammatory syndrome in mice. EMBO J. 2009, 28, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Singleton, P.A.; Bourguignon, L.Y.W.; Bikle, D.D. Calcium-induced human keratinocyte differentiation requires src- and fyn-mediated phosphatidylinositol 3-kinase–dependent activation of phospholipase c-γ1. Mol. Biol. Cell 2005, 16, 3236–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noto, P.B.; Bukhtiyarov, Y.; Shi, M.; McKeever, B.M.; McGeehan, G.M.; Lala, D.S. Regulation of sphingomyelin phosphodiesterase acid-like 3A gene (SMPDL3A) by liver X receptors. Mol. Pharmacol. 2012, 82, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Gazel, A.; Banno, T.; Walsh, R.; Blumenberg, M. Inhibition of JNK promotes differentiation of epidermal keratinocytes. J. Biol. Chem. 2006, 281, 20530–20541. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, T.; Wang, J.; Zhang, D.; Zhao, F.; Lin, X.; Wang, Z.; Shi, P.; Pang, X. miR-378b promotes differentiation of keratinocytes through nkx3.1. PLoS ONE 2015, 10, e0136049. [Google Scholar] [CrossRef]
- Bryja, A.; Dyszkiewicz-Konwińska, M.; Huang, Y.; Celichowski, P.; Nawrocki, M.J.; Jankowski, M.; Błochowiak, K.; Mehr, K.; Ramlau, P.; Nowicki, A.; et al. Genes involved in regulation of cellular metabolic processes, signaling and adhesion are the markers of porcine buccal pouch mucosal cells long-term primary cultured in vitro. J. Biol. Regul. Homeost. Agents 2018, 32, 1129–1141. [Google Scholar] [PubMed]
- Phatak, A.; Athar, M.; Crowell, J.A.; Leffel, D.; Herbert, B.S.; Bale, A.E.; Kopelovich, L. Crowell, David Leffel, Brittney-Shea Herbert, Allen, E. Bale and Levy Kopelovich Global gene expression of histologically normal primary skin cells from BCNS subjects reveals “single-hit” effects that are influenced by rapamycin. Oncotarget 2019, 10, 1360–1387. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, V.N.; Schon, M.P.; Seitz, C.S. c-Rel downregulation affects cell cycle progression of human keratinocytes. J. Investig. Dermatol. 2014, 134, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugasyan, R.; Voss, A.; Varigos, G.; Thomas, T.; Grumont, R.J.; Kaur, P.; Grigoriadis, G.; Gerondakis, S. The transcription factors c-rel and rela control epidermal development and homeostasis in embryonic and adult skin via distinct mechanisms. Mol. Cell. Biol. 2004, 24, 5733–5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Accession Number | Primer Sequence (5′–3′) | Product Size (bp) |
|---|---|---|---|
| CCL8 | NM_001164515.1 | CAATGGAAAGATCCCCTTCA CTCGCAGTCCAGGTAGGAAG | 206 |
| CXCL2 | NM_001001861.2 | CTGTGACCAAACGGAAGTCA AGCCAAATGCATGAAACACA | 237 |
| DACH1 | XM_001924267.6 | GGCATGGACAACCACTACCT CTTTTGTTCCATCGCCAACT | 233 |
| DUSP5 | XM_003359366 | TGCACGACCCACCTACACTA GCGAGATCACACTCCTCCTC | 250 |
| FABP5 | NM_001039746.2 | ATGGCAAAGACCTCACCATC CGAGTGCAGGTGACATTGTT | 244 |
| IL6 | NM_214399 | TGTCGAGGCTGTGCAGATTA GCATTTGTGGTGGGGTTAGG | 102 |
| PLK2 | XM_003133981 | AGCCTGCTTCCAGACAAAAA GAAGGAGGTAGAGCCGAGGT | 205 |
| PPARD | NM_001130241 | CAATGCCCTGGAACTCGATG TTGATCCGCTGCATCATCTG | 249 |
| PTGS2 | NM_214321 | AAAGGCCTCAATCGACCAGA ATCTGGGCGAGGCTTTTCTA | 202 |
| SPP1 | NM_214023.1 | ACTCCGATGAATCCGATGAG TCCGTCTCCTCACTTTCCAC | 220 |
| Gene | FC D7/D15 | FC D7/D30 | FC D15/D30 | p Value D7/D15 | p Value D7/D30 | p Value D15/D30 | Entez Gene ID |
|---|---|---|---|---|---|---|---|
| SPP1 | 0.0919949 | 0.0701564 | 0.762612 | 0.0162712 | 0.0230194 | 0.7663359 | 6696 |
| CCL8 | 0.1098375 | 0.1015572 | 0.9246133 | 0.0099092 | 0.002855 | 0.8648287 | 6355 |
| CXCL2 | 0.1585988 | 0.3618252 | 2.2813865 | 0.0282106 | 0.1000607 | 0.3277437 | 2920 |
| PLK2 | 0.1660003 | 0.6091754 | 3.6697255 | 0.0282106 | 0.3101504 | 0.1884621 | 10769 |
| DUSP5 | 0.1720052 | 0.3392398 | 1.9722648 | 0.0407438 | 0.1216767 | 0.4625691 | 1847 |
| PTGS2 | 0.176579 | 0.3277974 | 1.8563786 | 0.0332722 | 0.0861828 | 0.447703 | 5743 |
| SLC5A3 | 0.2252244 | 0.3389405 | 1.5049016 | 0.0282106 | 0.0555304 | 0.5402319 | 6526 |
| LIF | 0.252965 | 0.3537276 | 1.398326 | 0.0127508 | 0.0230194 | 0.3296551 | 3976 |
| CCL2 | 0.2544943 | 0.6892844 | 2.7084476 | 0.0407438 | 0.4043909 | 0.2350893 | 6347 |
| ATP1B1 | 0.2671037 | 0.4974017 | 1.8622043 | 0.0311185 | 0.1230303 | 0.3265278 | 481 |
| ATP13A3 | 0.2795739 | 0.291783 | 1.0436704 | 0.0338446 | 0.0406862 | 0.9731139 | 79572 |
| FABP5 | 0.2964051 | 0.2663389 | 0.8985637 | 0.0268357 | 0.0235015 | 0.8617697 | 2171 |
| GALNT7 | 0.3118498 | 0.5252929 | 1.6844421 | 0.0410788 | 0.1507523 | 0.415765 | 51809 |
| REL | 0.33281 | 0.5087624 | 1.5286872 | 0.0234279 | 0.0563739 | 0.3173859 | 5966 |
| ITGB3 | 0.3427886 | 0.7627618 | 2.2251672 | 0.038898 | 0.4213886 | 0.2350893 | 3690 |
| DACH1 | 0.3777917 | 0.3230132 | 0.8550034 | 0.0298064 | 0.0235639 | 0.7580772 | 1602 |
| SCARB1 | 0.3812346 | 0.5946187 | 1.5597186 | 0.0346365 | 0.1343271 | 0.3639803 | 949 |
| UGCG | 0.3838483 | 0.584988 | 1.5240083 | 0.0237712 | 0.0709872 | 0.283766 | 7357 |
| PDPN | 0.3878982 | 0.5660052 | 1.459159 | 0.0342294 | 0.1033121 | 0.4206462 | 10630 |
| LYN | 0.3967411 | 0.4074091 | 1.0268892 | 0.0346365 | 0.0423341 | 0.980695 | 4067 |
| ETS1 | 0.3978662 | 0.4650034 | 1.1687432 | 0.0311185 | 0.0474721 | 0.7511418 | 2113 |
| SMPDL3A | 0.4101801 | 0.3641437 | 0.8877656 | 0.0300194 | 0.0244065 | 0.8019706 | 10924 |
| PPARD | 0.4137917 | 0.2496635 | 0.6033556 | 0.0520061 | 0.0235015 | 0.3504653 | 5467 |
| FCER1G | 0.4253226 | 0.4158717 | 0.9777794 | 0.0268357 | 0.0235654 | 0.9761984 | 2207 |
| STEAP1 | 0.4256007 | 0.5300458 | 1.2454063 | 0.0268357 | 0.0442024 | 0.5192506 | 26872 |
| TGFB1 | 0.443799 | 0.8154499 | 1.8374307 | 0.0150164 | 0.2166007 | 0.0719885 | 7040 |
| RFC4 | 0.4463921 | 0.4260966 | 0.9545343 | 0.0346365 | 0.0362881 | 0.9445628 | 5984 |
| LMO2 | 0.4833797 | 0.5386858 | 1.1144154 | 0.0268357 | 0.0348947 | 0.744229 | 4005 |
| IL6 | 0.5575324 | 0.4445759 | 0.7973992 | 0.0884648 | 0.0442024 | 0.6294546 | 3569 |
| CEBPA | 0.8711062 | 0.4597589 | 0.5277875 | 0.3207823 | 0.017619 | 0.0452298 | 1050 |
| Cluster | Number of Cells | Share |
|---|---|---|
| Blue | 4641 | 76.33% |
| Red | 1320 | 21.71% |
| Green | 119 | 1.96% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryja, A.; Latosiński, G.; Jankowski, M.; Angelova Volponi, A.; Mozdziak, P.; Shibli, J.A.; Bryl, R.; Spaczyńska, J.; Piotrowska-Kempisty, H.; Krawiec, K.; et al. Transcriptomic and Morphological Analysis of Cells Derived from Porcine Buccal Mucosa—Studies on an In Vitro Model. Animals 2021, 11, 15. https://doi.org/10.3390/ani11010015
Bryja A, Latosiński G, Jankowski M, Angelova Volponi A, Mozdziak P, Shibli JA, Bryl R, Spaczyńska J, Piotrowska-Kempisty H, Krawiec K, et al. Transcriptomic and Morphological Analysis of Cells Derived from Porcine Buccal Mucosa—Studies on an In Vitro Model. Animals. 2021; 11(1):15. https://doi.org/10.3390/ani11010015
Chicago/Turabian StyleBryja, Artur, Grzegorz Latosiński, Maurycy Jankowski, Ana Angelova Volponi, Paul Mozdziak, Jamil A. Shibli, Rut Bryl, Julia Spaczyńska, Hanna Piotrowska-Kempisty, Krzysztof Krawiec, and et al. 2021. "Transcriptomic and Morphological Analysis of Cells Derived from Porcine Buccal Mucosa—Studies on an In Vitro Model" Animals 11, no. 1: 15. https://doi.org/10.3390/ani11010015
APA StyleBryja, A., Latosiński, G., Jankowski, M., Angelova Volponi, A., Mozdziak, P., Shibli, J. A., Bryl, R., Spaczyńska, J., Piotrowska-Kempisty, H., Krawiec, K., Kempisty, B., & Dyszkiewicz-Konwińska, M. (2021). Transcriptomic and Morphological Analysis of Cells Derived from Porcine Buccal Mucosa—Studies on an In Vitro Model. Animals, 11(1), 15. https://doi.org/10.3390/ani11010015