
Transcriptional Reprogramming in Rumen Epithelium during the Developmental Transition of Pre-Ruminant to the Ruminant in Cattle

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Tissue Collection
2.2. RNA Sequencing and Gene Expression Analysis
3. Results
3.1. RNA Sequencing and Transcriptomic Profiling of Rumen Epithelial Tissue
3.2. Impact of Weaning Transition on Transcriptome Atlas Dynamics in Rumen Epithelium
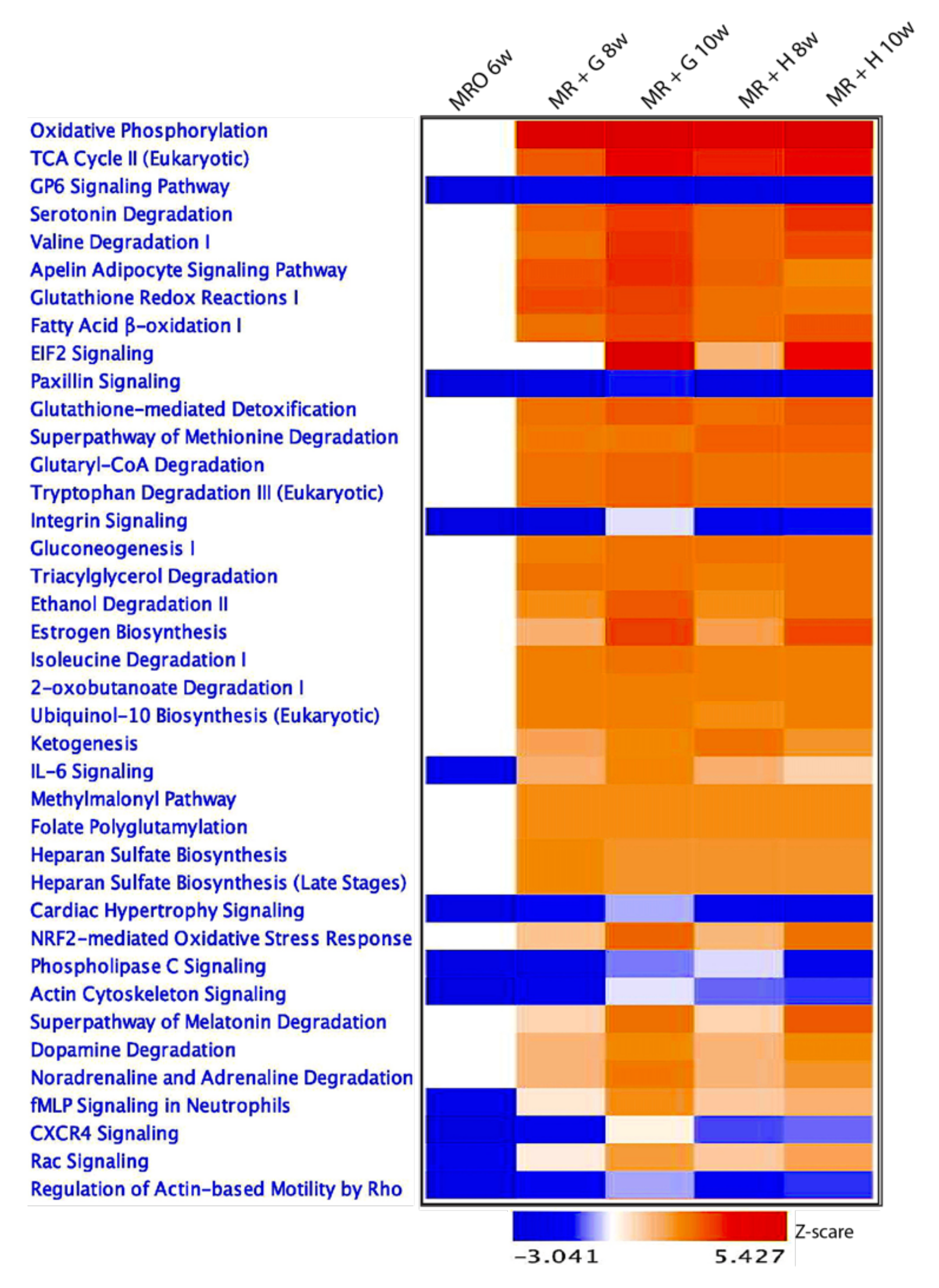
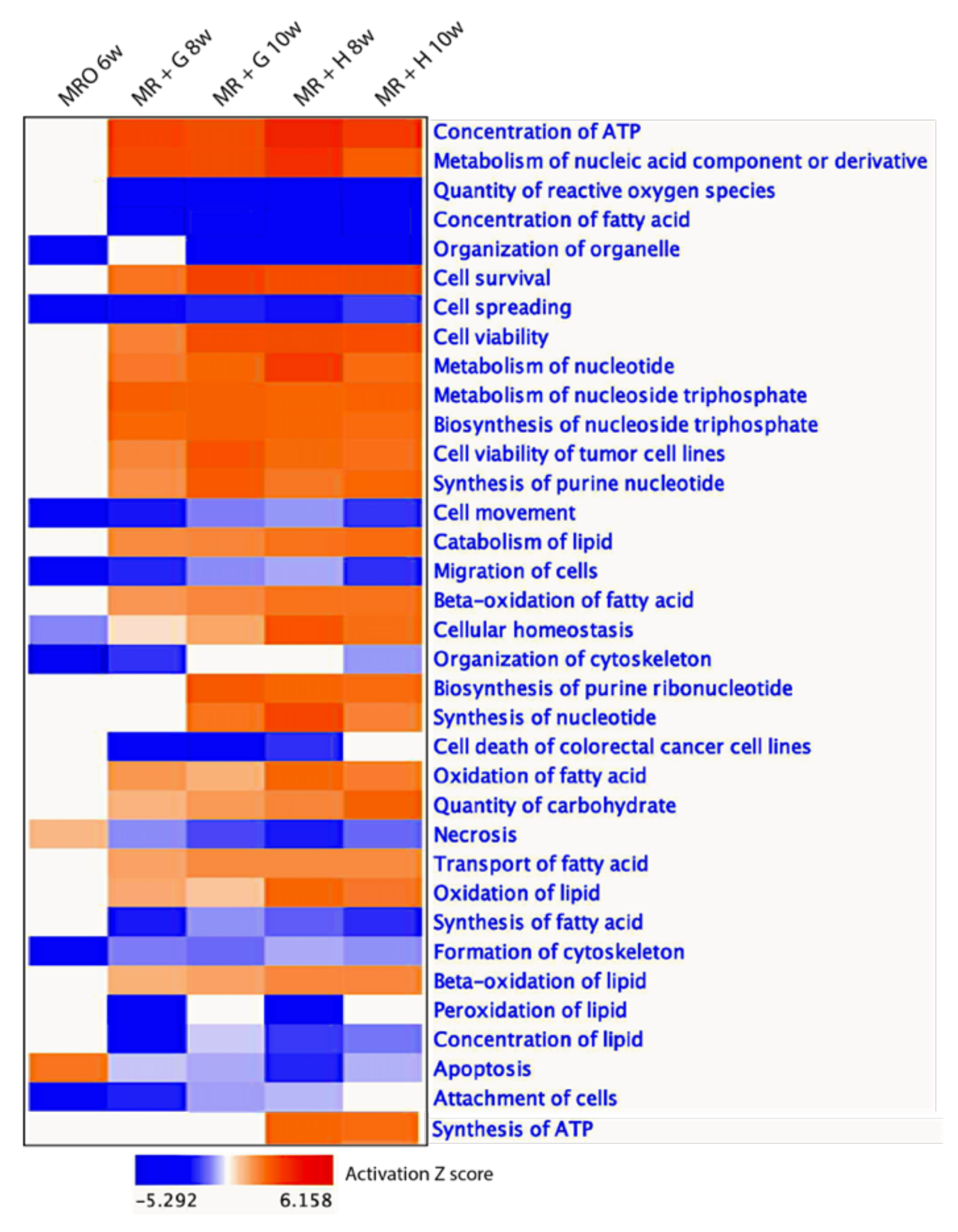
3.3. Transcriptional Reprogramming Induced by Weaning in Ruminal Epithelial Tissue and Reveals Significant Changes in Cellular Metabolism-Related Gene Regulation
3.4. The Developmental Shift of Gene Expression among the Temporal Groups during the Weaning Transition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malmuthuge, N.; Liang, G.; Guan, L.L. Regulation of rumen development in neonatal ruminants through microbial metagenomes and host transcriptomes. Genome Biol. 2019, 20, 172. [Google Scholar] [CrossRef] [Green Version]
- Diao, Q.; Zhang, R.; Fu, T. Review of Strategies to Promote Rumen Development in Calves. Animals 2019, 9, 490. [Google Scholar] [CrossRef] [Green Version]
- Steele, M.A.; Penner, G.B.; Chaucheyras-Durand, F.; Guan, L.L. Development and physiology of the rumen and the lower gut: Targets for improving gut health. J. Dairy Sci. 2016, 99, 4955–4966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meale, S.J.; Chaucheyras-Durand, F.; Berends, H.; Guan, L.L.; Steele, M.A. From pre- to postweaning: Transformation of the young calf’s gastrointestinal tract1. J. Dairy Sci. 2017, 100, 5984–5995. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L.; McLeod, K.R.; Capuco, A.V. Visceral tissue growth and proliferation during the bovine lactation cycle. J. Dairy Sci. 2004, 87, 2977–2986. [Google Scholar] [CrossRef] [Green Version]
- Baldwin Vi, R.L.; McLeod, K.R.; Klotz, J.L.; Heitmann, R.N. Rumen development, intestinal growth and hepatic metabolism in the pre- and postweaning ruminant. J. Dairy Sci. 2004, 87, E55–E65. [Google Scholar] [CrossRef] [Green Version]
- Biesheuvel, M.H.; Bijker, P.G.; Urlings, H.A. Some aspects of the gastrointestinal microflora of veal calves fed different rations: A pilot study. Vet. Q. 1991, 13, 97–104. [Google Scholar] [CrossRef]
- Khan, M.A.; Bach, A.; Weary, D.M.; von Keyserlingk, M.A.G. Invited review: Transitioning from milk to solid feed in dairy heifers. J. Dairy Sci. 2016, 99, 885–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, R.L.t.; Connor, E.E. Rumen Function and Development. Vet. Clin. N. Am. Food Anim. Pract. 2017, 33, 427–439. [Google Scholar] [CrossRef]
- Li, R.W.; Connor, E.E.; Li, C.; Baldwin Vi, R.L.; Sparks, M.E. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ. Microbiol. 2012, 14, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.-H. Transcriptional Reprogramming. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.-H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; pp. 2258–2259. [Google Scholar] [CrossRef]
- Himanen, S.V.; Sistonen, L. New insights into transcriptional reprogramming during cellular stress. J. Cell Sci. 2019, 132, jcs238402. [Google Scholar] [CrossRef] [PubMed]
- Renthal, W.; Tochitsky, I.; Yang, L.; Cheng, Y.-C.; Li, E.; Kawaguchi, R.; Geschwind, D.H.; Woolf, C.J. Transcriptional Reprogramming of Distinct Peripheral Sensory Neuron Subtypes after Axonal Injury. Neuron 2020, 108, 128–144.e129. [Google Scholar] [CrossRef]
- Connor, E.E.; Baldwin, R.L.t.; Li, C.J.; Li, R.W.; Chung, H. Gene expression in bovine rumen epithelium during weaning identifies molecular regulators of rumen development and growth. Funct. Integr. Genom. 2013, 13, 133–142. [Google Scholar] [CrossRef]
- Connor, E.E.; Siferd, S.; Elsasser, T.H.; Evock-Clover, C.M.; Van Tassell, C.P.; Sonstegard, T.S.; Fernandes, V.M.; Capuco, A.V. Effects of increased milking frequency on gene expression in the bovine mammary gland. BMC Genom. 2008, 9, 362. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, R.W.; Li, W.; Li, C.J. Transcriptome characterization by RNA-seq unravels the mechanisms of butyrate-induced epigenomic regulation in bovine cells. PLoS ONE 2012, 7, e36940. [Google Scholar] [CrossRef] [Green Version]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience 2020, 9, giaa021. [Google Scholar] [CrossRef] [Green Version]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. Jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [Green Version]
- Lever, J.; Krzywinski, M.; Altman, N. Principal component analysis. Nat. Methods 2017, 14, 641–642. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, R.L.t.; Li, R.W.; Jia, Y.; Li, C.J. Transcriptomic Impacts of Rumen Epithelium Induced by Butyrate Infusion in Dairy Cattle in Dry Period. Gene Regul. Syst. Biol. 2018, 12, 1177625018774798. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Fernandes, A.D.; Macklaim, J.M.; Linn, T.G.; Reid, G.; Gloor, G.B. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS ONE 2013, 8, e67019. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Rohr, K. Oxygen Is the High-Energy Molecule Powering Complex Multicellular Life: Fundamental Corrections to Traditional Bioenergetics. ACS Omega 2020, 5, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K.; et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015, 6, 8570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Weary, D.M.; Von Keyserlingk, M.A.G. Invited review: Effects of milk ration on solid feed intake, weaning, and performance in dairy heifers. J. Dairy Sci. 2011, 94, 1071–1081. [Google Scholar] [CrossRef]
- Yang, B.; Chen, H.; Cao, J.; He, B.; Wang, S.; Luo, Y.; Wang, J. Transcriptome Analysis Reveals That Alfalfa Promotes Rumen Development Through Enhanced Metabolic Processes and Calcium Transduction in Hu Lambs. Front. Genet. 2019, 10, 929. [Google Scholar] [CrossRef] [Green Version]
- Naeem, A.; Drackley, J.K.; Lanier, J.S.; Everts, R.E.; Rodriguez-Zas, S.L.; Loor, J.J. Ruminal epithelium transcriptome dynamics in response to plane of nutrition and age in young Holstein calves. Funct. Integr. Genom. 2014, 14, 261–273. [Google Scholar] [CrossRef]
- Wang, W.; Li, C.; Li, F.; Wang, X.; Zhang, X.; Liu, T.; Nian, F.; Yue, X.; Li, F.; Pan, X.; et al. Effects of early feeding on the host rumen transcriptome and bacterial diversity in lambs. Sci. Rep. 2016, 6, 32479. [Google Scholar] [CrossRef]
- Li, W.; Gelsinger, S.; Edwards, A.; Riehle, C.; Koch, D. Transcriptome analysis of rumen epithelium and meta-transcriptome analysis of rumen epimural microbial community in young calves with feed induced acidosis. Sci. Rep. 2019, 9, 4744. [Google Scholar] [CrossRef]
- Monsalve, M.; Wu, Z.; Adelmant, G.; Puigserver, P.; Fan, M.; Spiegelman, B.M. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol. Cell 2000, 6, 307–316. [Google Scholar] [CrossRef]
- Magri, L.; Swiss, V.A.; Jablonska, B.; Lei, L.; Pedre, X.; Walsh, M.; Zhang, W.; Gallo, V.; Canoll, P.; Casaccia, P. E2F1 coregulates cell cycle genes and chromatin components during the transition of oligodendrocyte progenitors from proliferation to differentiation. J. Neurosci. 2014, 34, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M.; Weiskirchen, R.; Breitkopf, K.; Dooley, S. Roles of TGF-beta in hepatic fibrosis. Front. Biosci. 2002, 7, d793–d807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Fang, L.; Kang, X.; Liu, S.; Liu, M.; Connor, E.E.; Baldwin, R.L.; Liu, G.; Li, C.J. Establishment and transcriptomic analyses of a cattle rumen epithelial primary cells (REPC) culture by bulk and single-cell RNA sequencing to elucidate interactions of butyrate and rumen development. Heliyon 2020, 6, e04112. [Google Scholar] [CrossRef]
- Lin, L.; Xie, F.; Sun, D.; Liu, J.; Zhu, W.; Mao, S. Ruminal microbiota-host crosstalk stimulates the development of the ruminal epithelium in a lamb model. Microbiota 2019, 7, 83. [Google Scholar] [CrossRef]
- Naeem, A.; Drackley, J.K.; Stamey, J.; Loor, J.J. Role of metabolic and cellular proliferation genes in ruminal development in response to enhanced plane of nutrition in neonatal Holstein calves. J. Dairy Sci. 2012, 95, 1807–1820. [Google Scholar] [CrossRef] [Green Version]
- Steele, M.A.; Vandervoort, G.; AlZahal, O.; Hook, S.E.; Matthews, J.C.; McBride, B.W. Rumen epithelial adaptation to high-grain diets involves the coordinated regulation of genes involved in cholesterol homeostasis. Physiol. Genom. 2011, 43, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Mentschel, J.; Leiser, R.; Mulling, C.; Pfarrer, C.; Claus, R. Butyric acid stimulates rumen mucosa development in the calf mainly by a reduction of apoptosis. Arch. Tierernahr. 2001, 55, 85–102. [Google Scholar] [CrossRef]
- Shen, H.; Xu, Z.; Shen, Z.; Lu, Z. The Regulation of Ruminal Short-Chain Fatty Acids on the Functions of Rumen Barriers. Front. Physiol. 2019, 10, 1305. [Google Scholar] [CrossRef]
- Wolin, M.J. The Rumen Fermentation: A Model for Microbial Interactions in Anaerobic Ecosystems. In Advances in Microbial Ecology: Volume 3; Alexander, M., Ed.; Springer: Boston, MA, USA, 1979; pp. 49–77. [Google Scholar] [CrossRef]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Liu, S.; Liu, M.; Kang, X.; Lin, S.; Li, B.; Connor, E.E.; Baldwin, R.L.t.; Tenesa, A.; Ma, L.; et al. Functional annotation of the cattle genome through systematic discovery and characterization of chromatin states and butyrate-induced variations. BMC Biol. 2019, 17, 68. [Google Scholar] [CrossRef]
- Nishihara, K.; Kato, D.; Suzuki, Y.; Kim, D.; Nakano, M.; Yajima, Y.; Haga, S.; Nakano, M.; Ishizaki, H.; Kawahara-Miki, R.; et al. Comparative transcriptome analysis of rumen papillae in suckling and weaned Japanese Black calves using RNA sequencing. J. Anim. Sci. 2018, 96, 2226–2237. [Google Scholar] [CrossRef]
- Connor, E.E.; Baldwin, R.L.; Walker, M.P.; Ellis, S.E.; Li, C.; Kahl, S.; Chung, H.; Li, R.W. Transcriptional regulators transforming growth factor-β1 and estrogen-related receptor-α identified as putative mediators of calf rumen epithelial tissue development and function during weaning1. J. Dairy Sci. 2014, 97, 4193–4207. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene 2002, 286, 81–89. [Google Scholar] [CrossRef]
- Puigserver, P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-alpha. Int. J. Obes. 2005, 29 (Suppl. 1), S5–S9. [Google Scholar] [CrossRef] [Green Version]
- Kimball, S.R. Eukaryotic initiation factor eIF2. Int. J. Biochem. Cell Biol. 1999, 31, 25–29. [Google Scholar] [CrossRef]
- Kim, Y.H.; Toji, N.; Kizaki, K.; Kushibiki, S.; Ichijo, T.; Sato, S. Effects of dietary forage and calf starter on ruminal pH and transcriptomic adaptation of the rumen epithelium in Holstein calves during the weaning transition. Physiol. Genom. 2016, 48, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Jablonowski, C.; Cheng, P.H.; AlTahan, A.; Li, C.; Wang, Y.; Palmer, L.; Lan, C.; Sun, B.; Abu-Zaid, A.; et al. KDM5A Regulates a Translational Program that Controls p53 Protein Expression. iScience 2018, 9, 84–100. [Google Scholar] [CrossRef] [Green Version]
- Sheaffer, K.L.; Kaestner, K.H. Transcriptional networks in liver and intestinal development. Cold Spring Harb. Perspect. Biol. 2012, 4, a008284. [Google Scholar] [CrossRef] [PubMed]
- Liefke, R.; Oswald, F.; Alvarado, C.; Ferres-Marco, D.; Mittler, G.; Rodriguez, P.; Dominguez, M.; Borggrefe, T. Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev. 2010, 24, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, P.; Zhang, L.; Bai, G.; Yang, C.; Wang, Y.; He, J.; Zhang, Z.; Zhu, G.; Zou, D. Superhero Rictor promotes cellular differentiation of mouse embryonic stem cells. Cell Death Differ. 2019, 26, 958–968. [Google Scholar] [CrossRef]
- Glidden, E.J.; Gray, L.G.; Vemuru, S.; Li, D.; Harris, T.E.; Mayo, M.W. Multiple site acetylation of Rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J. Biol. Chem. 2012, 287, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jebali, A.; Dumaz, N. The role of RICTOR downstream of receptor tyrosine kinase in cancers. Mol. Cancer 2018, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Verdoni, A.M.; Ikeda, S.; Ikeda, A. Serum response factor is essential for the proper development of skin epithelium. Mamm. Genome 2010, 21, 64–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyngaarden, J.B. Regulation of purine biosynthesis and turnover. Adv. Enzym. Regul. 1976, 14, 25–42. [Google Scholar] [CrossRef]
- Li, C.J.; Li, R.W.; Wang, Y.H.; Elsasser, T.H. Pathway analysis identifies perturbation of genetic networks induced by butyrate in a bovine kidney epithelial cell line. Funct. Integr. Genom. 2007, 7, 193–205. [Google Scholar] [CrossRef]
- Li, C.J.; Li, R.W.; Baldwin, R.L., VI. Assembly and Analysis of Changes in Transcriptomes of Dairy Cattle Rumen Epithelia during Lactation and Dry Periods. Agric. Sci. 2018, 9, 619–638. [Google Scholar] [CrossRef] [Green Version]
- Petri, R.M.; Wetzels, S.U.; Qumar, M.; Khiaosa-Ard, R.; Zebeli, Q. Adaptive responses in short-chain fatty acid absorption, gene expression, and bacterial community of the bovine rumen epithelium recovered from a continuous or transient high-grain feeding. J. Dairy Sci. 2019, 102, 5361–5378. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA-seq Statistics | Mean ± STD |
|---|---|
| Average Reads per sample | 44,888,781 ± 5,226,700 |
| Mapped pairs % | 91.0 ± 2.4 |
| Mapped to gene % | 83.98 ± 0.53 |
| Mapped to Intergenic % | 16.02 ± 0.53 |
| Forward % of reads mapped | 50.37 ± 0.13 |
| Reverse % of reads mapped | 49.63 ± 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldwin VI, R.L.; Liu, M.; Connor, E.E.; Ramsay, T.G.; Liu, G.E.; Li, C.-J. Transcriptional Reprogramming in Rumen Epithelium during the Developmental Transition of Pre-Ruminant to the Ruminant in Cattle. Animals 2021, 11, 2870. https://doi.org/10.3390/ani11102870
Baldwin VI RL, Liu M, Connor EE, Ramsay TG, Liu GE, Li C-J. Transcriptional Reprogramming in Rumen Epithelium during the Developmental Transition of Pre-Ruminant to the Ruminant in Cattle. Animals. 2021; 11(10):2870. https://doi.org/10.3390/ani11102870
Chicago/Turabian StyleBaldwin VI, Ransom L., Mei Liu, Erin E. Connor, Timothy G. Ramsay, George E. Liu, and Cong-Jun Li. 2021. "Transcriptional Reprogramming in Rumen Epithelium during the Developmental Transition of Pre-Ruminant to the Ruminant in Cattle" Animals 11, no. 10: 2870. https://doi.org/10.3390/ani11102870