
Lactation Associated Genes Revealed in Holstein Dairy Cows by Weighted Gene Co-Expression Network Analysis (WGCNA)



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. Milk Composition Detection
2.4. RNA Extraction and cDNA Library Analysis
2.5. Weighted Gene Coexpression Network Analysis
2.5.1. Gene Coexpression Network Construction
2.5.2. Interaction Analysis of Coexpression Modules
2.5.3. Module–Trait Relationships Analysis
2.6. Sequencing Data Validation by qRT-PCR
2.7. Statistical Analysis
3. Results
3.1. Milk Yield and Component Results
3.2. Transcriptome Sequencing Data Analysis
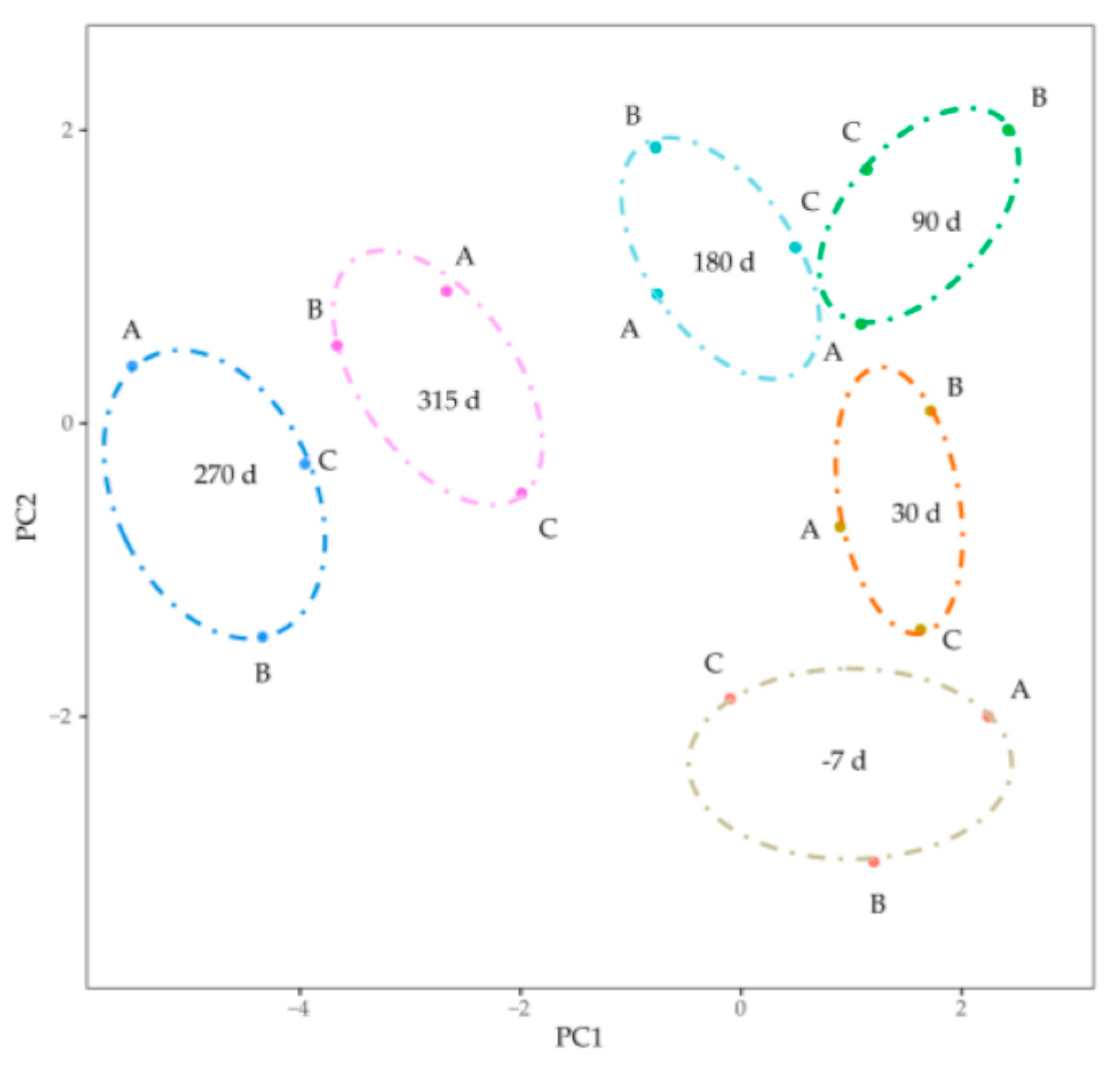
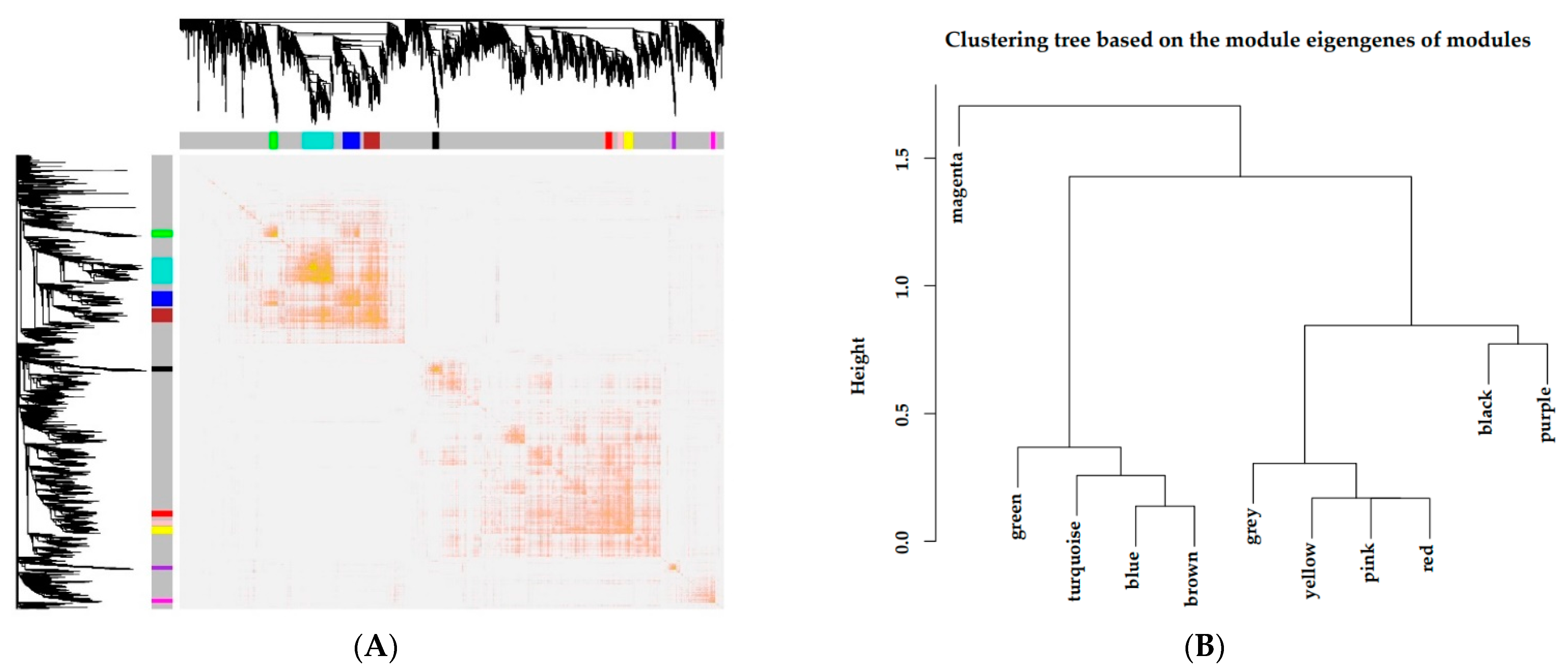
3.3. Gene Coexpression Network Construction
3.4. Interaction Analysis of Coexpression Modules
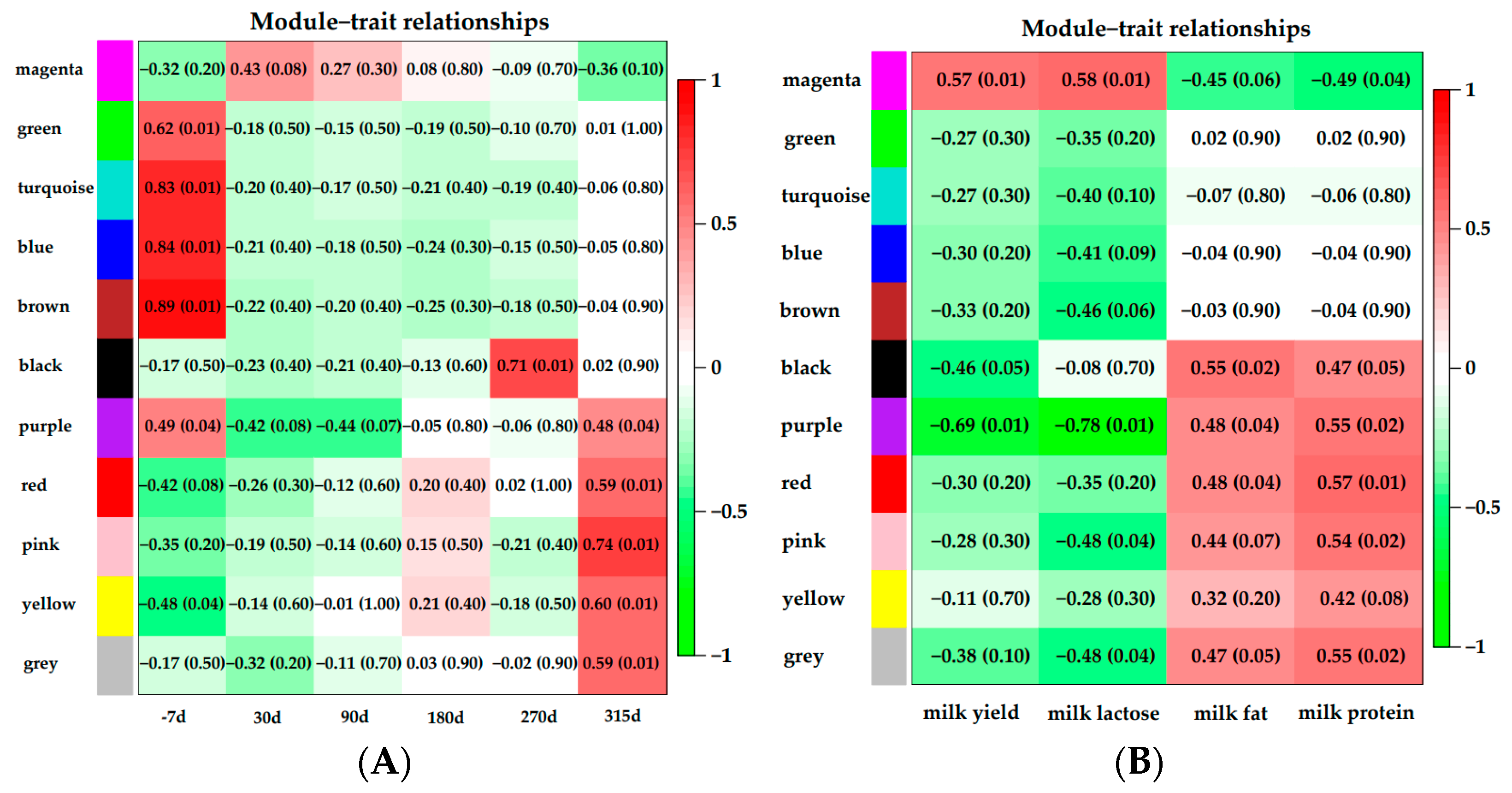
3.5. Module–Trait Relationships Analysis
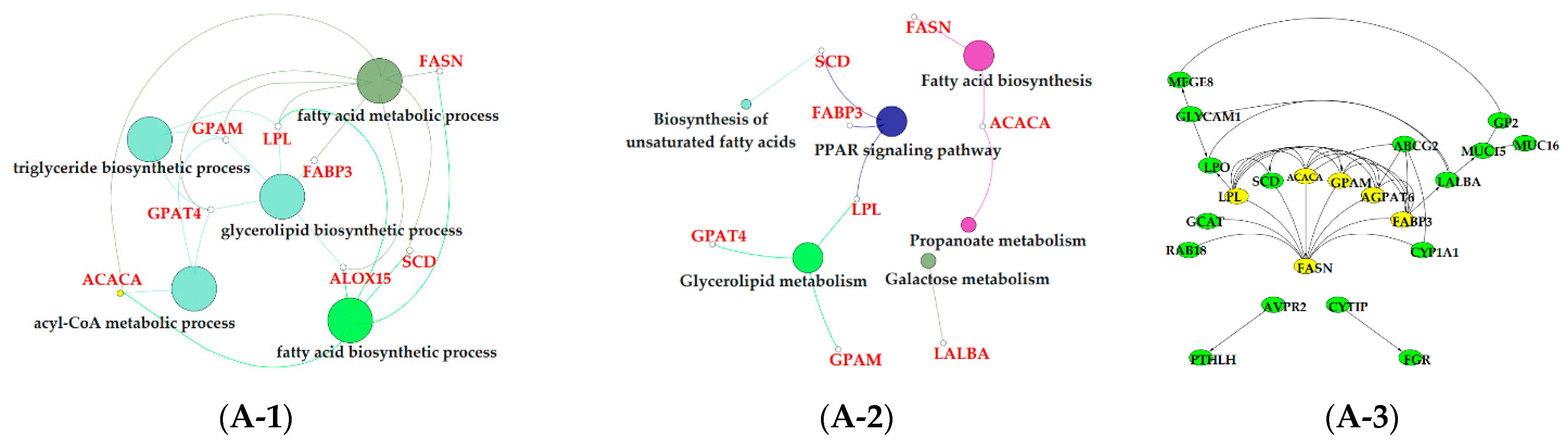
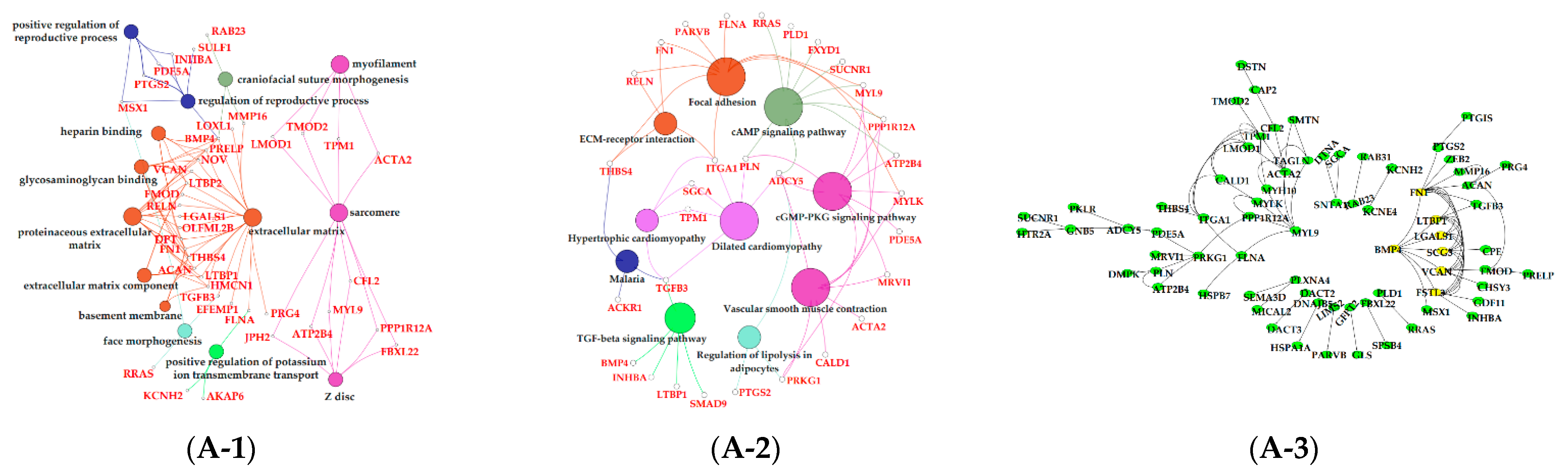
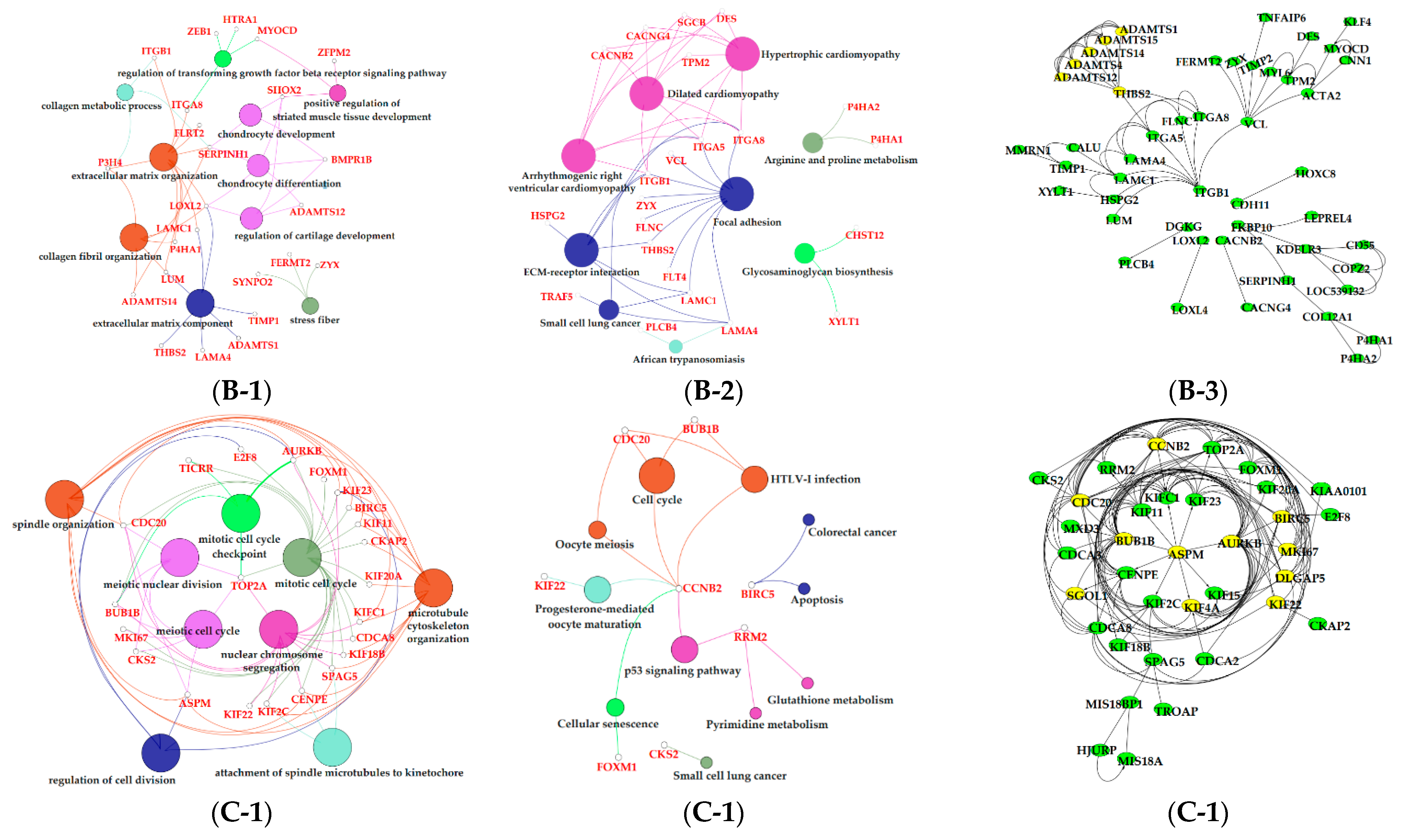
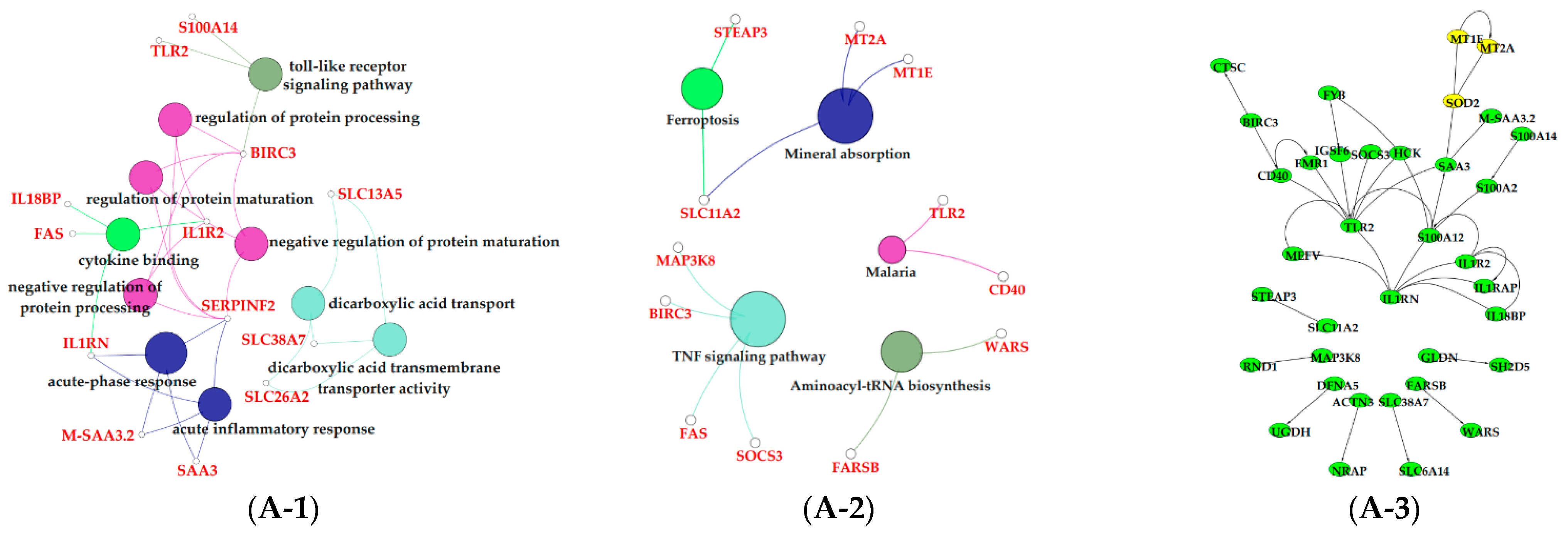
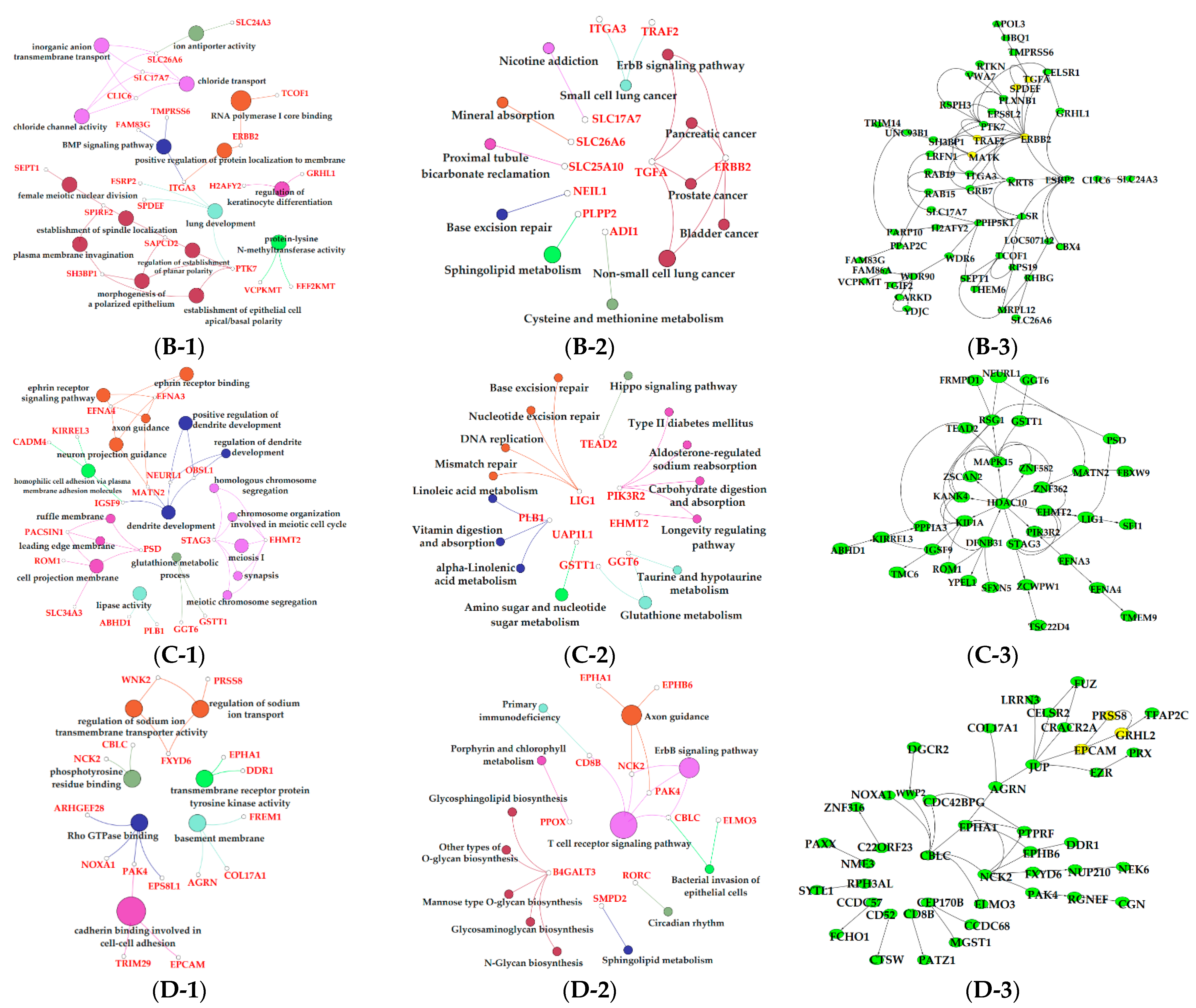
3.6. Functional Enrichment Analysis of Critical Modules
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Y.; Fan, D.; Hang, F.; Yan, B.; Zhao, J.; Zhang, H.; Chen, W. Effect of Calcium on Absorption Properties and Thermal Stability of Milk during Microwave Heating. Int. J. Mol. Sci. 2018, 19, 1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Liu, J.; Huang, B.; Kan, X.; Chen, G.; Wang, W.; Fu, S. Kisspeptin-10 Induces β-Casein Synthesis via GPR54 and Its Downstream Signaling Pathways in Bovine Mammary Epithelial Cells. Int. J. Mol. Sci. 2017, 18, 2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Liu, Z.; Chao, T.; Hou, L.; Fan, R.; He, R.; Wang, G.; Wang, J. Screening of miRNA profiles and construction of regulation networks in early and late lactation of dairy goat mammary glands. Sci. Rep. 2017, 7, 11933. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, J.; Chaudhury, M.; Dang, A.K. Alterations in the milk yield and composition during different stages of lactation cycle in elite and non-elite Karan-Fries cross-bred cows (Holstein Fresian x Tharparkar). Biol. Rhythm Res. 2017, 48, 499–506. [Google Scholar] [CrossRef]
- Sherchand, L.; Mcnew, R.W.; Kellogg, D.W.; Johnson, Z.B. Selection of a Mathematical Model to Generate Lactation Curves Using Daily Milk Yields of Holstein Cows. J. Dairy Sci. 1995, 78, 2507–2513. [Google Scholar] [CrossRef]
- Hossein-Zadeh, N.G. Application of growth models to describe the lactation curves for test-day milk production in Holstein cows. J. Appl. Anim. Res. 2016, 45, 145–151. [Google Scholar] [CrossRef]
- Gao, Y.; Lin, X.; Shi, K.; Yan, Z.; Wang, Z. Bovine mammary gene expression profiling during the onset of lactation. PLoS ONE 2013, 8, e70393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.T.; Zou, Y.X.; White, R.R.; Liu, J.X.; Liu, H.Y. Transcriptomic profiles of the bovine mammary gland during lactation and the dry period. Funct. Integr. Genom. 2018, 18, 125–140. [Google Scholar] [CrossRef]
- Lin, Y.; Lv, H.; Jiang, M.; Zhou, J.; Song, S.; Hou, X. Functional analysis of the dairy cow mammary transcriptome between early lactation and mid-dry period. J. Dairy Res. 2019, 86, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Bionaz, M.; Loor, J.J. Gene networks driving bovine milk fat synthesis during the lactation cycle. BMC Genom. 2008, 9, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bionaz, M.; Loor, J.J. Gene networks driving bovine mammary protein synthesis during the lactation cycle. Bioinform. Biol. Insights 2011, 5, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Finucane, K.A.; McFadden, T.B.; Bond, J.P.; Kennelly, J.J.; Zhao, F.Q. Onset of lactation in the bovine mammary gland: Gene expression profiling indicates a strong inhibition of gene expression in cell proliferation. Funct. Integr. Genom. 2008, 8, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Bionaz, M.; Periasamy, K.; Rodriguez-Zas, S.L.; Everts, R.E.; Lewin, H.A.; Hurley, W.L.; Loor, J.J. Old and new stories: Revelations from functional analysis of the bovine mammary transcriptome during the lactation cycle. PLoS ONE 2012, 7, e33268. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhang, L.; Wang, H.; Lu, J.; Wei, H.; Yu, S. Transcriptomic Profiling of Young Cotyledons Response to Chilling Stress in Two Contrasting Cotton (Gossypium hirsutum L.) Genotypes at the Seedling Stage. Int. J. Mol. Sci. 2020, 21, 5095. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, B.; Tang, X.; Zhang, J.; Lin, J. Comparative Transcriptome Analysis of Different Dendrobium Species Reveals Active Ingredients-Related Genes and Pathways. Int. J. Mol. Sci. 2020, 21, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, Y.; Zheng, Q.; Sui, M.; Zhu, L.; Xu, L.; Zhang, Y.; Liu, Y.; Fang, F.; Chu, M.; Ma, Y.; et al. Comprehensive Analysis of LncRNA Reveals the Temporal-Specific Module of Goat Skeletal Muscle Development. Int. J. Mol. Sci. 2019, 20, 3950. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Cheng, H.; Soomro, M.; Shuyan, L.; Bilal Tufail, M.; Nazir, M.F.; Feng, X.; Zhang, Y.; Dongyun, Z.; Limin, L.; et al. Comparative Transcriptomic Analysis to Identify the Genes Related to Delayed Gland Morphogenesis in Gossypium bickii. Genes 2020, 11, 472. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, C.; Xu, C.; Sun, J.; Grierson, D.; Zhang, B.; Chen, K. Integration of Metabolite Profiling and Transcriptome Analysis Reveals Genes Related to Volatile Terpenoid Metabolism in Finger Citron (C. medica var. sarcodactylis). Molecules 2019, 24, 2564. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Sun, B.; Mi, X.; Xiao, Z. Gene co-expression network for analysis of plasma exosomal miRNAs in the elderly as markers of aging and cognitive decline. PeerJ 2020, 8, e8318. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, F.; Luo, S.; Yin, X.; He, D.; Liu, J.; Yue, Z.; Song, J. Co-expression of key gene modules and pathways of human breast cancer cell lines. Biosci. Rep. 2019, 39, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Piantoni, P.; Wang, P.; Drackley, J.K.; Hurley, W.L.; Loor, J.J. Expression of metabolic, tissue remodeling, oxidative stress, and inflammatory pathways in mammary tissue during involution in lactating dairy cows. Bioinform. Biol. Insights 2010, 4, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, M.; Cai, D.; Hao, Y.; Zhao, X.; Zhu, Y.; Zhu, H.; Yang, Z. Composition, coagulation characteristics, and cheese making capacity of yak milk. J. Dairy Sci. 2020, 103, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Rivetti, C.; Tauler, R.; Piña, B.; Barata, C. Tryptophan hydroxylase (TRH) loss of function mutations in Daphnia deregulated growth, energetic, serotoninergic and arachidonic acid metabolic signalling pathways. Sci. Rep. 2019, 9, 3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Chen, W.; Yao, Q.; Guo, B.; Valliyodan, B.; Wang, Z.; Nguyen, H.T. Genome-wide transcriptional profiling for elucidating the effects of brassinosteroids on Glycine max during early vegetative development. Sci. Rep. 2019, 9, 16085. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.J.; Zhao, Q.Q.; Liu, X.S.; Dong, S.H.; E, J.F.; Li, X.; Liu, C.; Wang, H. Toll-like receptor 4 regulates spontaneous intestinal tumorigenesis by up-regulating IL-6 and GM-CSF. J. Cell. Mol. Med. 2020, 24, 385–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Xu, F.; Gu, L.; Rong, G.; Li, M.; Qiao, F.; Shi, L.; Wang, D.; Xia, W.; Xun, W.; et al. Landscape of alternative splicing in Capra_hircus. Sci. Rep. 2018, 8, 15128. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Gao, X.; Ding, J.; Liu, C.; Du, C.; Hou, C.; Zhu, J.; Lou, B. Transcriptome Sequencing Reveals the Traits of Spermatogenesis and Testicular Development in Large Yellow Croaker (Larimichthys crocea). Genes 2019, 10, 958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.; Choi, J.; Lee, G.W.; Park, S.Y.; Huh, A.; Dean, R.A.; Lee, Y.H. Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci. Rep. 2015, 24, 8567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Ramsköld, D.; Wang, E.T.; Burge, C.B.; Sandberg, R. An Abundance of Ubiquitously Expressed Genes Revealed by Tissue Transcriptome Sequence Data. PLoS Comput. Biol. 2009, 5, e1000598. [Google Scholar] [CrossRef]
- Trakhtenberg, E.F.; Pho, N.; Holton, K.M.; Chittenden, T.W.; Goldberg, J.L.; Dong, L. Cell types differ in global coordination of splicing and proportion of highly expressed genes. Sci. Rep. 2016, 31, 32249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, T.; Li, K.; Zheng, P.; Wang, Y.; Lv, Y.; Shen, L.; Chen, Y.; Xue, Z.; Li, B.; Jin, L.; et al. Weighted Gene Coexpression Network Analysis Identified MicroRNA Coexpression Modules and Related Pathways in Type 2 Diabetes Mellitus. Oxidative Med. Cell. Longev. 2019, 2019, 9567641. [Google Scholar] [CrossRef] [PubMed]
- Foroughi, K.; Amini, M.; Atashi, A.; Mahmoodzadeh, H.; Hamann, U.; Manoochehri, M. Tissue-Specific Down-Regulation of the Long Non-Coding RNAs PCAT18 and LINC01133 in Gastric Cancer Development. Int. J. Mol. Sci. 2018, 19, 3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Li, T.; Qin, S.; Yu, S.; Chu, Q.; Li, A.; Wu, K. Identifying Tumorigenesis and Prognosis-Related Genes of Lung Adenocarcinoma: Based on Weighted Gene Coexpression Network Analysis. BioMed Res. Int. 2020, 2020, 4169691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, B.L.; Zhang, L.J.; Gao, L.; Ma, F.C.; He, R.Q.; Chen, G.; Ma, J.; Zhong, J.C.; Hu, X.H. Downregulation of miR-224-5p in prostate cancer and its relevant molecular mechanism via TCGA, GEO database and in silico analyses. Oncol. Rep. 2018, 40, 3171–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backman, M.; Flenkenthaler, F.; Blutke, A.; Dahlhoff, M.; Ländström, E.; Renner, S.; Philippou-Massier, J.; Krebs, S.; Rathkolb, B.; Prehn, C.; et al. Multi-omics insights into functional alterations of the liver in insulin-deficient diabetes mellitus. Mol. Metab. 2019, 26, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wang, Q.; Zhou, W.; Liu, T.; Yang, L.; Zheng, P.; Zhang, L.; Ji, G. Integrated analysis of hepatic mRNA and miRNA profiles identified molecular networks and potential biomarkers of NAFLD. Sci. Rep. 2018, 8, 7628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Fan, L.; Xu, X.; Fu, J.; Xue, Y. Identification of core genes and pathways in type 2 diabetes mellitus by bioinformatics analysis. Mol. Med. Rep. 2019, 20, 2597–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Zhao, Y.; Zou, Q.; Fang, X.; Peng, S. Bioinformatics applications on Apache Spark. GigaScience 2018, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, D.; Liu, S.; Yuan, T.; Guo, J.; Fang, L.; Du, G. Systematic Elucidation of the Mechanism of Genistein against Pulmonary Hypertension via Network Pharmacology Approach. Int. J. Mol. Sci. 2019, 20, 5569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sun, Y.; Cole, J.R. A scalable and accurate targeted gene assembly tool (SAT-Assembler) for next-generation sequencing data. PLoS Comput. Biol. 2014, 10, e1003737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Langfelder, P.; Fuller, T.; Dong, J.; Li, A.; Hovarth, S. Weighted gene coexpression network analysis: State of the art. J. Biopharm. Stat. 2010, 20, 281–300. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sun, X.; Hou, X.; Qu, B.; Gao, X.; Li, Q. Effects of glucose on lactose synthesis in mammary epithelial cells from dairy cow. BMC Vet. Res. 2016, 12, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, A.; Aris, A.; Guasch, I. Consequences of supplying methyl donors during pregnancy on the methylome of the offspring from lactating and non-lactating dairy cattle. PLoS ONE 2017, 12, e0189581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powe, C.E.; Puopolo, K.M.; Newburg, D.S.; Lönnerdal, B.; Chen, C.; Allen, M.; Merewood, A.; Worden, S.; Welt, C.K. Effects of recombinant human prolactin on breast milk composition. Pediatrics 2011, 127, e359–e366. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Cai, W.; Zhou, C.; Yin, H.; Zhang, Z.; Loor, J.J.; Sun, D.; Zhang, Q.; Liu, J.; Zhang, S. RNA-Seq reveals 10 novel promising candidate genes affecting milk protein concentration in the Chinese Holstein population. Sci. Rep. 2016, 2, 26813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.; Lee, H.J.; Kim, K.; Caetano-Anolles, K.; Jeong, J.Y.; Park, S.; Oh, Y.K.; Cho, S.; Kim, H. Characterizing Milk Production Related Genes in Holstein Using RNA-seq. Asian-Australas. J. Anim. Sci. 2016, 29, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Capuco, A.V.; Ellis, S.E.; Hale, S.A.; Long, E.; Erdman, R.A.; Zhao, X.; Paape, M.J. Lactation persistency: Insights from mammary cell proliferation studies. J. Anim. Sci. 2003, 81, 18–31. [Google Scholar] [CrossRef] [Green Version]
- Capuco, A.V.; Wood, D.L.; Baldwin, R.; McLeod, K.; Paape, M.J. Mammary cell number, proliferation, and apoptosis during a bovine lactation: Relation to milk production and effect of bST. J. Dairy Sci. 2001, 84, 2177–2187. [Google Scholar] [CrossRef]
- Marti, A.; Lazar, H.; Ritter, P.; Jaggi, R. Transcription factor activities and gene expression during mouse mammary gland involution. J. Mammary Gland. Biol. Neoplasia 1999, 4, 145–152. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Vaillant, F.; Alexander, W.S.; Wintermantel, T.M.; Forrest, N.C.; Holroyd, S.L.; McManus, E.J.; Schutz, G.; Watson, C.J.; Chodosh, L.A.; et al. c-myc as a mediator of accelerated apoptosis and involution in mammary glands lacking Socs3. EMBO J. 2006, 25, 5805–5815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton, S.; Keenan, T.W. The milk fat globule membrane. Biochim. Biophys. Acta 1975, 415, 273–309. [Google Scholar] [CrossRef]
- El-Loly, M.M. Composition, Properties and Nutritional Aspects of Milk Fat Globule Membrane—A Review. Pol. J. Food Nutr. Ences 2011, 61, 7–32. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Xia, H.; Jiang, J.; Xu, X.; Li, M.; Chen, Z.; Sun, Y.; Zhang, H.; Yang, Z. MicroRNA-212 targets SIRT2 to influence lipogenesis in bovine mammary epithelial cell line. J. Dairy Res. 2020, 87, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Klonoff, D.C. Replacements for trans fats-will there be an oil shortage? J. Diabetes Sci. Technol. 2007, 1, 415–422. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Days | 30 d | 90 d | 180 d | 270 d | SEM | p |
|---|---|---|---|---|---|---|
| Milk yield (Kg) | 32.65 b | 34.40 a | 31.12 c | 26.46 d | 0.13 | <0.0001 |
| Milk lactose (%) | 5.05 b | 5.11 a | 5.04 b | 4.93 c | 0.01 | 0.0007 |
| Milk fat (%) | 3.40 d | 3.48 c | 3.58 b | 3.87 a | 0.02 | <0.0001 |
| Milk protein (%) | 3.12 c | 3.15 c | 3.24 b | 3.32 a | 0.01 | <0.0001 |
| Module Colors | Red | Purple | Green | Black | Yellow | Pink | Blue | Turquoise | Brown | Magenta | Grey |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Node Number | 63 | 38 | 75 | 62 | 83 | 50 | 164 | 286 | 151 | 41 | 12,163 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Arbab, A.A.I.; Zhang, H.; Yang, Y.; Nazar, M.; Han, Z.; Yang, Z. Lactation Associated Genes Revealed in Holstein Dairy Cows by Weighted Gene Co-Expression Network Analysis (WGCNA). Animals 2021, 11, 314. https://doi.org/10.3390/ani11020314
Fan Y, Arbab AAI, Zhang H, Yang Y, Nazar M, Han Z, Yang Z. Lactation Associated Genes Revealed in Holstein Dairy Cows by Weighted Gene Co-Expression Network Analysis (WGCNA). Animals. 2021; 11(2):314. https://doi.org/10.3390/ani11020314
Chicago/Turabian StyleFan, Yongliang, Abdelaziz Adam Idriss Arbab, Huimin Zhang, Yi Yang, Mudasir Nazar, Ziyin Han, and Zhangping Yang. 2021. "Lactation Associated Genes Revealed in Holstein Dairy Cows by Weighted Gene Co-Expression Network Analysis (WGCNA)" Animals 11, no. 2: 314. https://doi.org/10.3390/ani11020314