
Habitat Elevation Shapes Microbial Community Composition and Alter the Metabolic Functions in Wild Sable (Martes zibellina) Guts

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Sites
2.2. Fecal Sample Collection
2.3. DNA Extraction
2.4. 16S rRNA Gene Amplification and Sequencing
2.5. DNA-Based qPCR Assay
2.6. Bioinformatics
2.7. Statistical Analysis
3. Results
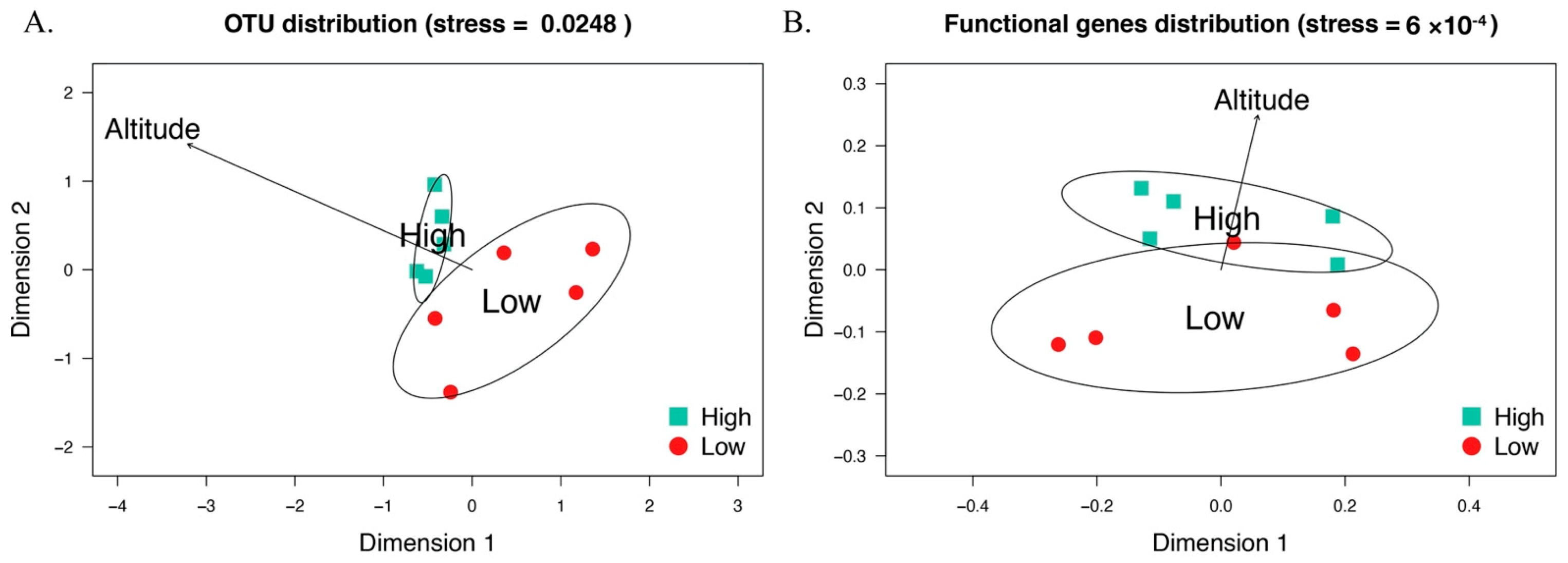
3.1. Microbial Diversity and Community Composition in Sable Gut
3.2. Function Profiles of Sable Intestinal Microbiome
4. Discussion
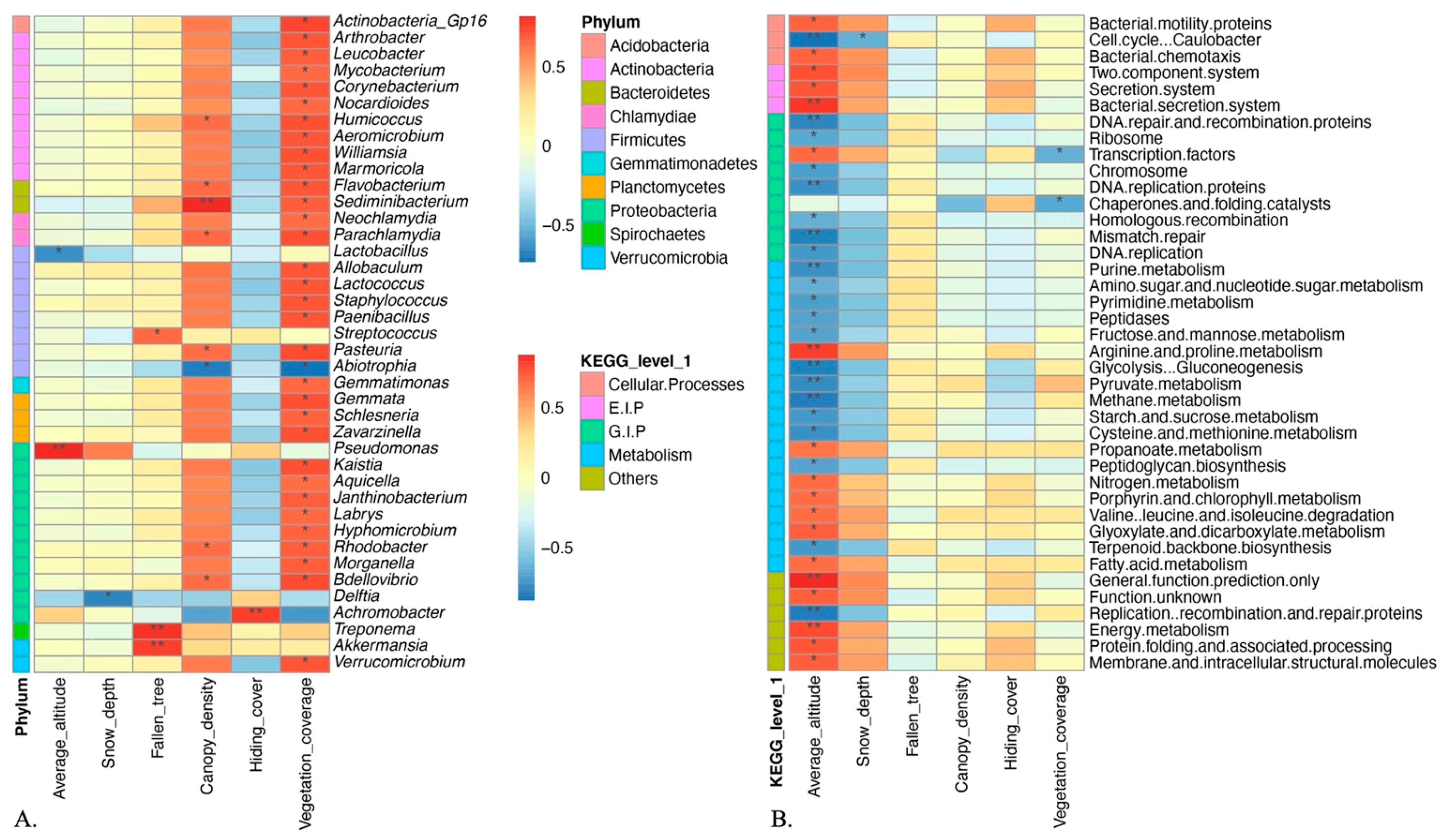
4.1. Environmental Factors Can Influence Sable Gut Microbial Community
4.2. Gut Bacterial Structure Varies Along Altitude Increase
4.3. Changes of Gut Microbial Function in Cold Area
4.4. Wild Sable Survives in Harsh Environment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aubry, K.B. Biology and Conservation of Martens, Sables, and Fishers. Northwest Sci. 2001, 63, 192. [Google Scholar]
- Aubry, K.B.; Zielinski, W.J.; Raphael, M.G.; Proulx, G.; Buskirk, S.W. Biology and Conservation of Martens, Sables, and Fishers (A New Synthesis) || 19. Conservation of Martens, Sables, and Fishers in Multispecies Bioregional Assessments; Cornell University Press: Ithaca, NY, USA, 2017. [Google Scholar]
- Wang, S. China Red Data Book of Endangered Animals; Science Press: Beijing, China, 1998. [Google Scholar]
- Buskirk, S.W.; Ma, Y.; Xu, L. Sable Ecology in Chinese taiga forests. Res. Explor. 1993, 9, 479–480. [Google Scholar]
- Zakharov, E.S.; Safronov, V.M. Ecology of sable of North-Western Yakutian native population. Russ. J. Theriol. 2017, 16, 74–85. [Google Scholar] [CrossRef]
- Baillie, J.E.M.; Hilton, C.; Stuart, S.N. IUCN Red List of Threatened Species: A Global Species Assessment; IUCN-The World Conservation Union: Gland, Switzerland, 2004. [Google Scholar]
- Li, Z. Wildlife Protection Law of the People’s Republic of China; Committee of the National People’s Congress: Beijing, China, 2018.
- Murakami, T. Food habits of the Japanese sable Martes zibellina brachyura in eastern Hokkaido, Japan. Mamm. Study 2003, 28, 129–134. [Google Scholar] [CrossRef]
- Kashtanov, S.N.; Stolpovsky, Y.A.; Meshchersky, I.G.; Svishcheva, G.R.; Veprev, S.G.; Somova, M.M.; Shitova, M.V.; Meshchersky, S.I.; Rozhnov, V.V. Taxonomic status and genetic identification of Altai sable (Martes zibellina averini Bazhanov, 1943). Russ. J. Genet. 2018, 54, 1342–1351. [Google Scholar] [CrossRef]
- Monakhov, V.G. Morphological peculiarities as indicators of natural history of sable (Martes zibellina) in North-Asia Pacific coast. Russ. J. Ecol. 2016, 47, 493–500. [Google Scholar] [CrossRef]
- Monakhov, V.G. Martes zibellina (Carnivora: Mustelidae). Mamm. Species 2011, 876, 75–86. [Google Scholar] [CrossRef]
- Pulliainen, E. Food and feeding habits of the pine marten in Finnish forest lapland in winter. Worldw. Furbear. Conf. Proc. 1981, 1, 580–598. [Google Scholar]
- Buskirk, S.W. Seasonal food habits of marten in south-central Alaska. J. Wildl. Manag. 1984, 48, z84–z133. [Google Scholar] [CrossRef]
- Buskirk, S.W.; Ma, Y.; Xu, L.; Jiang, Z. Diets of, and prey selection by, sables (Martes zibellina) in Northern China. J. Mammal. 1996, 77, 725–730. [Google Scholar] [CrossRef]
- Mustonen, A.M.; Puukka, M.; Saarela, S.; Paakkonen, T.; Aho, J.; Nieminen, P. Adaptations to fasting in a terrestrial mustelid, the sable (Martes zibellina). Comp. Biochem. Phys. A 2006, 144, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Proulx, G.; Aubry, K.; Birks, J.; Buskirk, S.; Zielinski, W. World Distribution and Status of the Genus Martes in 2000. In Martens and Fishers (Martes) in Human-Altered Environments; Springer: New York, NY, USA, 2004. [Google Scholar]
- Zhang, R.; Yang, L.; Ai, L.; Yang, Q.; Chen, M. Geographic characteristics of sable (Martes zibellina) distribution over time in Northeast China. Ecol. Evol. 2017, 7, 4016–4023. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, N.; Tang, X.; Liu, N.; Zhao, W. Changes in intestinal microbiota across an altitudinal gradient in the lizard Phrynocephalus vlangalii. Ecol. Evol. 2018, 8, 4695–4703. [Google Scholar] [CrossRef] [Green Version]
- Jin, G. Comparison of Wintering Habitat Selection and Habitat Suitability of Sable between the Changbai Mountains and the Greater Khingan Mountains in Northeastern China; Northeast Forestry University: Harbin, China, 2016. [Google Scholar]
- Guo, K.; Liu, H.; Bao, H.; Hu, J.; Wang, S.; Zhang, W.; Zhao, Y.; Jiang, G. Habitat selection and their interspecific interactions for mammal assemblage in the Greater Khingan Mountains, northeastern China. Wildl. Biol. 2017, 1, wlb.00261. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, C.; Stojanović, O.; Colin, D.J.; Suarez-Zamorano, N.; Tarallo, V.; Veyrat-Durebex, C.; Rigo, D.; Fabbiano, S.; Stevanović, A.; Hagemann, S.; et al. Gut microbiota orchestrates energy homeostasis during cold. Cell 2015, 163, 1360–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J.I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Chen, Y.H.; Chuang, H.L.; Chiu, C.C.; Huang, C.C. Investigation of the effects of microbiota on exercise physiological adaption, performance, and energy utilization using a gnotobiotic animal model. Front. Microbiol. 2019, 10, 13. [Google Scholar] [CrossRef]
- Bragg, M.; Freeman, E.W.; Lim, H.C.; Songsasen, N.; Muletz-Wolz, C.R. Gut microbiomes differ among dietary types and stool consistency in the captive red wolf (Canis rufus). Front. Microbiol. 2020, 11, 11. [Google Scholar] [CrossRef]
- David, L.; Maurice, C.; Carmody, R.; Gootenberg, D.; Button, J.; Wolfe, B.; Ling, A.; Devlin, A.; Varma, Y.; Fischbach, M. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Vital, M.; Gao, J.; Rizzo, M.; Harrison, T.; Tiedje, J.M. Diet is a major factor governing the fecal butyrate-producing community structure across Mammalia, Aves and Reptilia. ISME J. 2014, 9, 832–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tang, H.; Zhang, C.; Zhao, Y.; Derrien, M.; Rocher, E.; Van-Hylckama Vlieg, J.E.; Strissel, K.; Zhao, L.; Obin, M. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J. 2015, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Round, L.J.; Mazmanian, K.S. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 600. [Google Scholar] [CrossRef]
- Sun, C.H.; Liu, H.Y.; Liu, B.; Yuan, B.D.; Lu, C.H. Analysis of the gut microbiome of wild and captive Pere David′s deer. Front. Microbiol. 2019, 10, 9. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Rex Gaskins, H.; Stumpf, R.M.; Yildirim, S.; Torralba, M. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- Qin, W.; Song, P.; Lin, G.; Huang, Y.; Wang, L.; Zhou, X.; Li, S.; Zhang, T. Gut Microbiota plasticity influences the adaptability of wild and domestic animals in co-inhabited areas. Front. Microbiol. 2020, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Switzer, A.; Watanabe-Chailland, M.; Bik, E.M.; Relman, D.A.; Romick-Rosendale, L.E.; Ollberding, N.J. Reduced gut microbiome diversity and metabolome differences in rhinoceros species at risk for iron overload disorder. Front. Microbiol. 2019, 10, 15. [Google Scholar] [CrossRef]
- Suzuki, M.T.; Giovannoni, S.J. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 1996, 62, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.H.; Yao, Y.F.; Dong, M.M.; Xia, T.R.; Li, D.Y.; Xie, M.; Wu, J.Y.; Wen, A.X.; Wang, Q.; Zhu, G.X.; et al. Characterisation of the gut microbial community of rhesus macaques in high-altitude environments. BMC Microbiol. 2020, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, H.Y.; Li, L.M.; Zhang, X.J.; Chen, J.P. The effect of disease and season to hepatopancreas and intestinal mycobiota of Litopenaeus vannamei. Front. Microbiol. 2019, 10, 13. [Google Scholar] [CrossRef]
- Bestion, E.; Jacob, S.; Zinger, L.; Gesu, L.D.; Richard, M.; White, J.; Cote, J. Climate warming reduces gut microbiota diversity in a vertebrate ectotherm. Nat. Ecol. Evol. 2017, 1, 161. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.E.; Hauffe, H.C.; Bull, M.J.; Atwood, T.C.; Perkins, S.E. Global change-driven use of onshore habitat impacts polar bear faecal microbiota. ISME J. 2019, 13, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teyssier, A.; Matthysen, E.; Hudin, N.S.; de Neve, L.; White, J.; Lens, L. Diet contributes to urban-induced alterations in gut microbiota: Experimental evidence from a wild passerine. Proc. R. Soc. B Boil. Sci. 2020, 287, 20192182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, Y.; Shi, Q.; Peng, Z.; Hou, Z. The Gastrointestinal Parasites and Bacteria Community of Sable (Mates zibellina) in Northeastern China. Pak. J. Zool. 2018, 50, 50. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, H.; Gao, X.; Shang, S.; Wu, X.; Chen, J.; Zhang, W.; Zhang, W.; Jiang, M.; Zhang, B. Comparison of the bacterial communities in feces from wild versus housed sables (Martes zibellina) by high-throughput sequence analysis of the bacterial 16S rRNA gene. AMB Express 2016, 6, 98. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Hui, N.; Simpanen, S.; Tudeer, L.; Romantschuk, M. Simulation of microbial response to accidental diesel spills in basins containing brackish sea water and sediment. Front. Microbiol. 2020, 11, 593232. [Google Scholar] [CrossRef]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.K.; Schmidt, T.M. rrnDB: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Soonvald, L.; Loit, K.; Runno-Paurson, E.; Astover, A.; Tedersoo, L. The role of long-term mineral and organic fertilisation treatment in changing pathogen and symbiont community composition in soil. Appl. Soil Ecol. 2019, 141, 45–53. [Google Scholar] [CrossRef]
- Huse, S.M.; Welch, D.M.; Morrison, H.G.; Sogin, M.L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 2010, 12, 1889–1898. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Wang, Q. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.P.; Veach, A.M.; Rigdon-Huss, A.R.; Grond, K.; Lickteig, S.K.; Lothamer, K.; Oliver, A.K.; Jumpponen, A. Scraping the bottom of the barrel: Are rare high throughput sequences artifacts? Fungal Ecol. 2015, 13, 221–225. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. GigaScience 2012, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; Mcdonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.; Kindt, R.; Legendre, P.; O′Hara, R.; Simpson, G.; Solymos, P.; Stevens, M.; Wagner, H. Package Vegan: Community Ecology Package; Time International: Ames, IA, USA, 2012. [Google Scholar] [CrossRef]
- Ginestet, C. ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2012, 174, 245–246. [Google Scholar] [CrossRef]
- Suzuki, T.A.; Martins, F.M.; Nachman, M.W. Altitudinal variation of the gut microbiota in wild house mice. Mol. Ecol. 2018, 28, 2378–2390. [Google Scholar] [CrossRef]
- Huang, B.H.; Chang, C.W.; Huang, C.W.; Jian, G.; Liao, P.C. Composition and functional specialists of the gut microbiota of frogs reflect habitat differences and agricultural activity. Front. Microbiol. 2018, 8, 2670. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, A.; Hui, N.; Puhakka, R.; Oikarinen, S.; Gronroos, M.; Selonen, V.A.O.; Siter, N.; Kramna, L.; Roslund, M.I.; Vari, H.K.; et al. Yard vegetation is associated with gut microbiota composition. Sci. Total Environ. 2020, 713, 8. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Mishra, S.; Zhao, J.C.; Tang, J.S.; Zeng, B.; Kong, F.L.; Ning, R.H.; Li, M.; Zhang, H.Z.; Zeng, Y.T.; et al. Metagenomic study suggests that the gut microbiota of the giant panda (Ailuropoda melanoleuca) may not be specialized for fiber fermentation. Front. Microbiol. 2018, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.H.; Wang, X.; Bernstein, S.; Huffman, M.A.; Xia, D.P.; Gu, Z.Y.; Chen, R.; Sheeran, L.K.; Wagner, R.S.; Li, J.H. Marked variation between winter and spring gut microbiota in free-ranging Tibetan Macaques (Macaca thibetana). Sci. Rep. 2016, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Zeng, B.; Zhang, S.Y.; Xu, H.L.; Kong, F.L.; Yu, X.Q.; Wang, P.; Yang, M.Y.; Li, D.Y.; Zhang, M.W.; Ni, Q.Y.; et al. Gut microbiota of Tibetans and Tibetan pigs varies between high and low altitude environments. Microbiol. Res. 2020, 235, 9. [Google Scholar] [CrossRef]
- Li, H.; Li, T.; Beasley, D.E.; Petr, H.; Xiao, Z.; Zhang, S.; Li, J.; Lin, Q.; Li, X. Diet diversity is associated with beta but not alpha diversity of pika gut microbiota. Front. Microbiol. 2016, 7, 1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Li, Y.; Sun, J.; Guo, Y. Studies on the Antagonistic Properties of Lactobacillus Acidophilus in the Imitative Gastroenteric Environment. Microbiol. China 2002, 4, 35–38. [Google Scholar] [CrossRef]
- Alquati, C.; Gioia, L.D.; Santarossa, G.; Alberghina, L.; Lotti, M. The cold-active lipase of Pseudomonas fragi. Eur. J. Biochem. 2002, 269, 3321–3328. [Google Scholar] [CrossRef]
- Wang, W.; Wang, F.; Li, L.; Wang, A.; Sharshov, K.; Druzyaka, A.; Lancuo, Z.; Wang, S.; Shi, Y. Characterization of the gut microbiome of black-necked cranes (Grus nigricollis) in six wintering areas in China. Arch. Microbiol. 2020, 202, 983–993. [Google Scholar] [CrossRef]
- Pandey, A.; Jain, R.; Sharma, A.; Dhakar, K.; Kaira, G.S.; Rahi, P.; Dhyani, A.; Pandey, N.; Adhikari, P.; Shouche, Y.S. 16S rRNA gene sequencing and MALDI-TOF mass spectrometry based comparative assessment and bioprospection of psychrotolerant bacteria isolated from high altitudes under mountain ecosystem. SN Appl. Sci. 2019, 1, 278. [Google Scholar] [CrossRef] [Green Version]
- Cheviron, Z.A.; Bachman, G.C.; Connaty, A.D.; McClelland, G.B.; Storz, J.F. Regulatory changes contribute to the adaptive enhancement of thermogenic capacity in high-altitude deer mice. Proc. Natl. Acad. Sci. USA 2012, 109, 8635–8640. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Zhang, H.; Wei, Q.; Zhao, C.; Yang, X.; Wu, X.; Xia, T.; Liu, G.; Zhang, L.; Gao, Y. Comparative analyses of fecal microbiota in European mouflon (Ovis orientalis musimon) and blue sheep (Pseudois nayaur) living at low or high Altitudes. Front. Microbiol. 2019, 10, 1735. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Iskender, A.U.; Srikanth, K.; Kim, H.; Zhunushov, A.T.; Chooq, H.; Jang, G.W.; Lim, Y.; Song, K.D.; Park, J.E. Transcriptome of chicken liver tissues reveals the candidate genes and pathways responsible for adaptation into two different climatic Conditions. Animals 2019, 9, 1076. [Google Scholar] [CrossRef] [Green Version]
- Raza, M.F.; Wang, Y.C.; Cai, Z.H.; Bai, S.; Yao, Z.C.; Awan, U.A.; Zhang, Z.Y.; Zheng, W.W.; Zhang, H.Y. Gut microbiota promotes host resistance to low-temperature stress by stimulating its arginine and proline metabolism pathway in adult Bactrocera dorsalis. PLoS Pathog. 2020, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.H.; Guan, G.B.; Dai, Y.; Cao, C.J.; Tao, L.; Du, H.; Nobile, C.J.; Zhong, J.; Huang, G.H. Lactic acid bacteria differentially regulate filamentation in two heritable cell types of the human fungal pathogen Candida albicans. Mol. Microbiol. 2016, 102, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Zhao, X.Q.; Han, X.P.; Xu, S.X.; Zhao, L.; Hu, L.Y.; Xu, T.W.; Zhao, N.; Zhang, X.L.; Chen, D.D.; et al. Comparative study of gut microbiota in Tibetan wild asses (Equus kiang) and domestic donkeys (Equus asinus) on the Qinghai-Tibet plateau. PeerJ 2020, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhu, J.; Chen, J.; Ding, H.; Gao, Y.; Jiang, W.; Yuan, K.; Dong, X.; Chen, L. Characterization of snow leopard (Panthera uncia) facal microbiota by barcoded pyrosequencing. Chin. J. Wildl. 2019, 873–881. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Altitude | Snow Depth (cm) | Vegetation Types | Fallen Tree | Canopy Density | Hiding Cover | Vegetation Coverage |
|---|---|---|---|---|---|---|---|
| Sable01 | Low a | 15 | Mixed c | 0 | 0.2 | 0.7 | 0.3 |
| Sable02 | High b | 20 | Mixed | 2 | 0.6 | 0.4 | 0.8 |
| Sable03 | Low | 20 | Broad d | 2 | 0.5 | 0.4 | 0.6 |
| Sable05 | Low | 30 | Mixed | 1 | 0.3 | 0.7 | 0.5 |
| Sable07 | High | 35 | Mixed | 0 | 0.4 | 0.7 | 0.5 |
| Sable08 | High | 25 | Broad | 2 | 0.4 | 0.8 | 0.5 |
| Sable11 | High | 25 | Mixed | 0 | 0.3 | 0.8 | 0.5 |
| Sable12 | Low | 20 | Broad | 1 | 0.2 | 0.3 | 0.3 |
| Sable13 | Low | 20 | Broad | 1 | 0.3 | 0.7 | 0.4 |
| Sable16 | High | 30 | Broad | 5 | 0.5 | 0.7 | 0.6 |
| Phyla | Environmental Factors | p | r |
|---|---|---|---|
| Firmicutes | Average altitude | 0.033 | −0.67 |
| Proteobacteria | Average altitude | 0.009 | 0.77 |
| Spirochaetes | Fallen wood | 0.004 | 0.82 |
| Deferribacteres | Fallen wood | 0.042 | 0.65 |
| Verrucomicrobia | Vegetation Coverage | 0.036 | 0.66 |
| Gemmatimonadetes | Vegetation Coverage | 0.040 | 0.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, L.; Liu, X.; Jin, G.; Ma, Y.; Tan, H.; Khalid, M.; Romantschuk, M.; Yin, S.; Hui, N. Habitat Elevation Shapes Microbial Community Composition and Alter the Metabolic Functions in Wild Sable (Martes zibellina) Guts. Animals 2021, 11, 865. https://doi.org/10.3390/ani11030865
Su L, Liu X, Jin G, Ma Y, Tan H, Khalid M, Romantschuk M, Yin S, Hui N. Habitat Elevation Shapes Microbial Community Composition and Alter the Metabolic Functions in Wild Sable (Martes zibellina) Guts. Animals. 2021; 11(3):865. https://doi.org/10.3390/ani11030865
Chicago/Turabian StyleSu, Lantian, Xinxin Liu, Guangyao Jin, Yue Ma, Haoxin Tan, Muhammed Khalid, Martin Romantschuk, Shan Yin, and Nan Hui. 2021. "Habitat Elevation Shapes Microbial Community Composition and Alter the Metabolic Functions in Wild Sable (Martes zibellina) Guts" Animals 11, no. 3: 865. https://doi.org/10.3390/ani11030865