
Successful and Unsuccessful Brain Aging in Pets: Pathophysiological Mechanisms behind Clinical Signs and Potential Benefits from Palmitoylethanolamide Nutritional Intervention


Abstract
:Simple Summary
Abstract
1. Successful Aging and the Brain
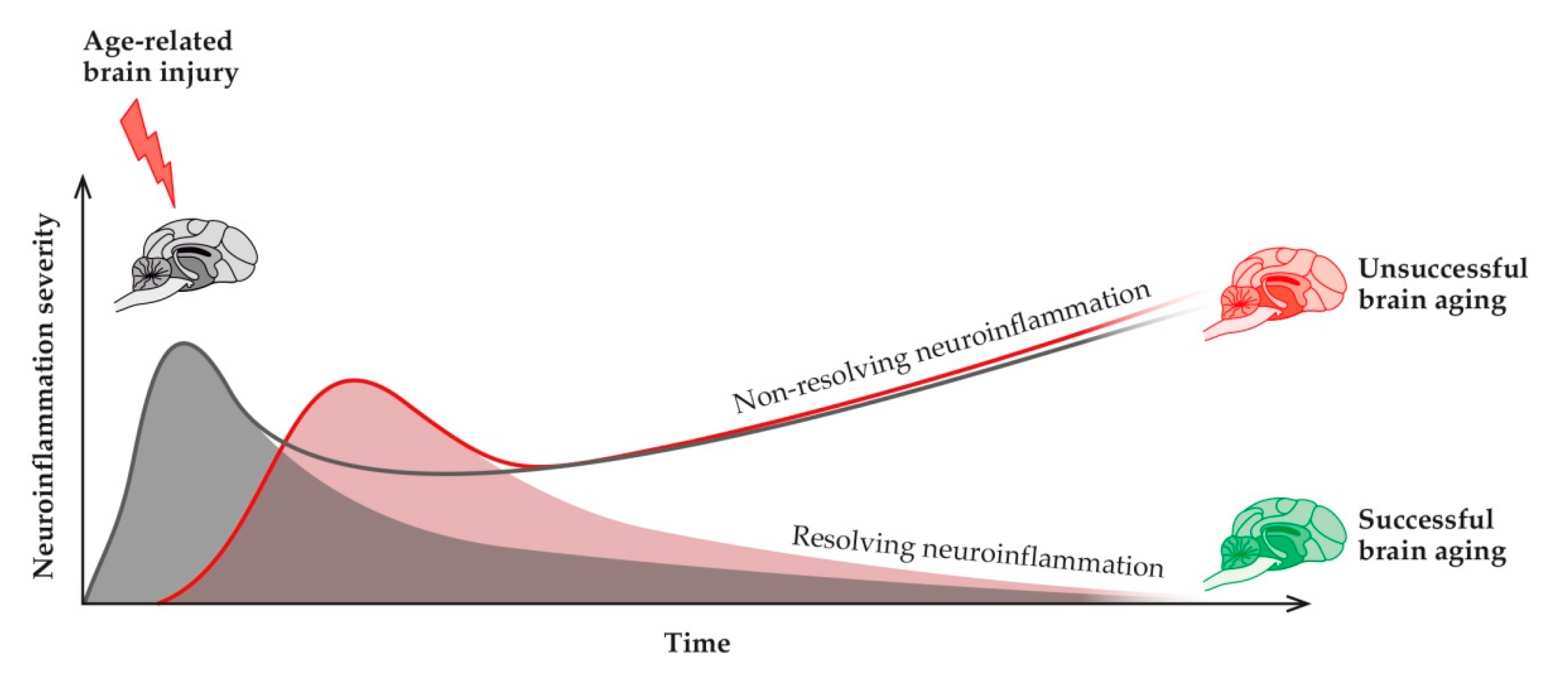
2. Unsuccessful Brain Aging in Pets
3. Neurobehavioral and Physical Signs
4. Diagnostic Considerations
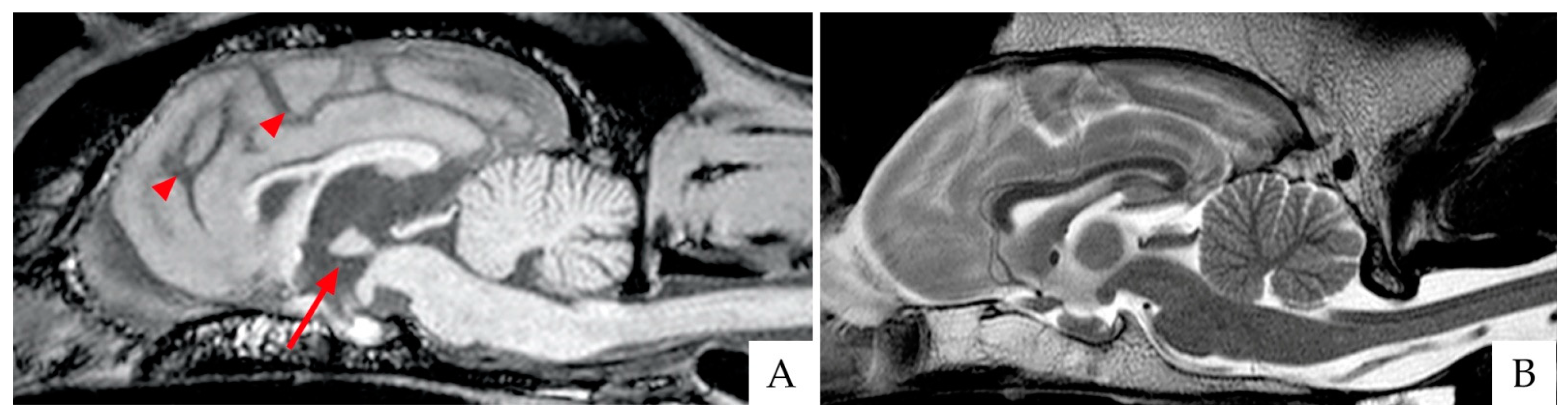
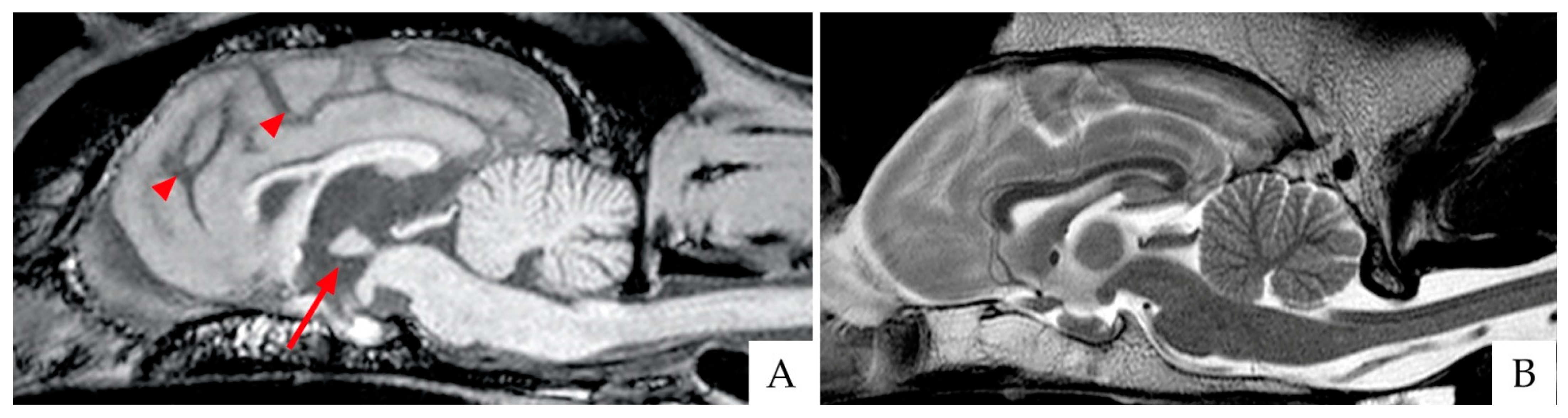
4.1. Brain Imaging
4.2. Putative Biomarkers
5. Prevalence and Disease Progression
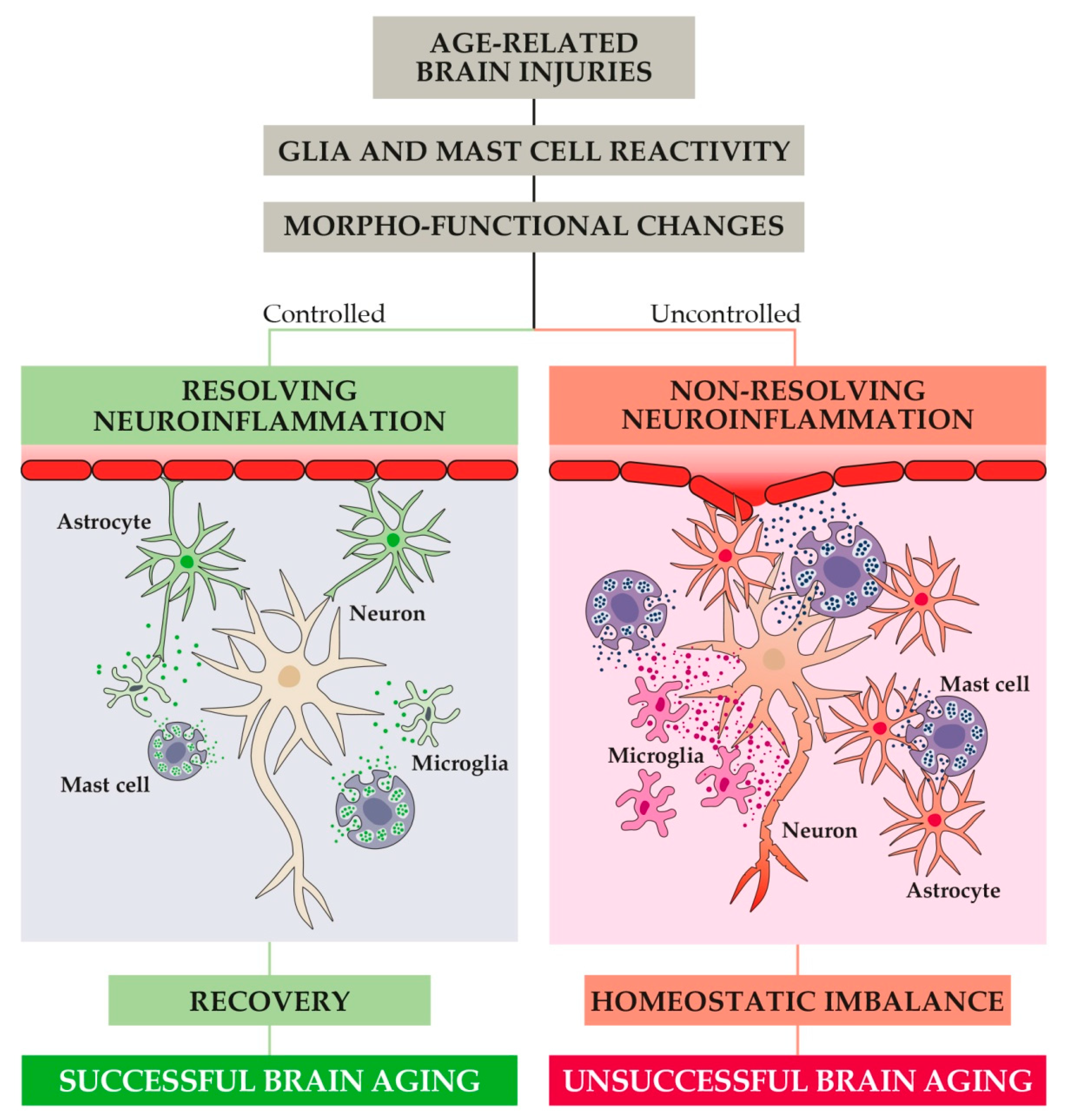
6. Pathological Features
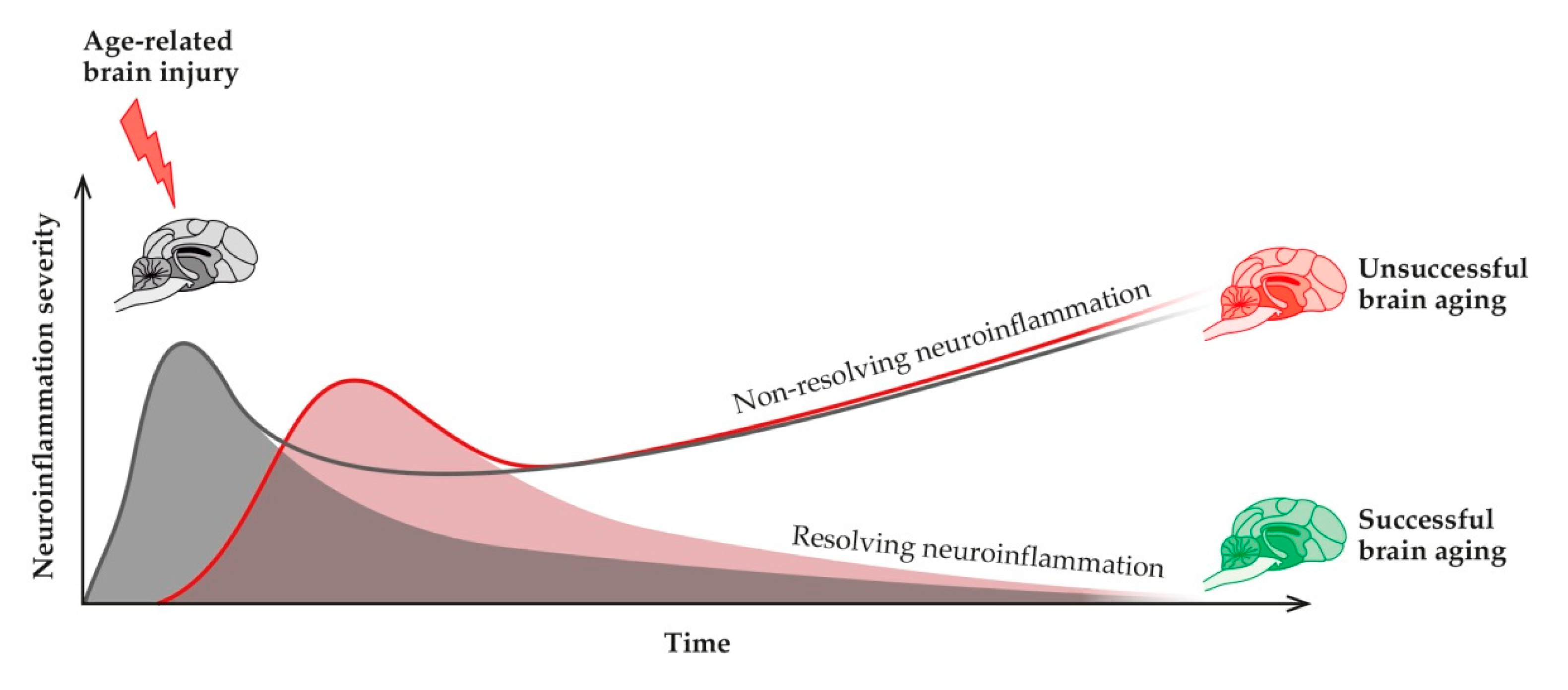
7. The Neuroinflammatory Process and Its Role in Healthy and Pathological Aging
7.1. Neuroinflammation
7.2. Neuroinflammation in Alzheimer’s Disease
8. The Role of Astrocytes, Microglia, and CNS Mast Cells in Alzheimer’s Disease
9. Pro-Resolving Mediators in the Resolution of Age-Related Neuroinflammation
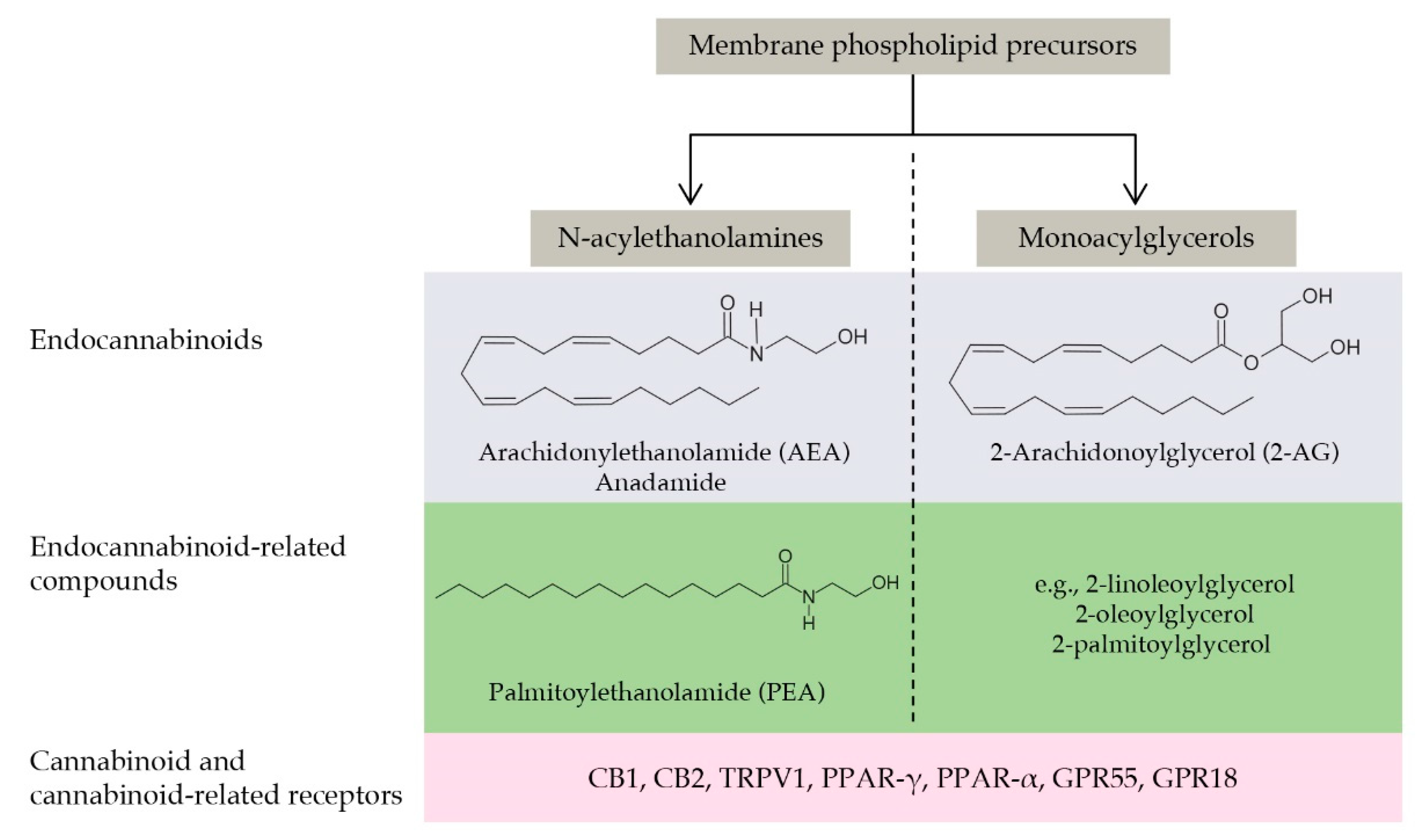
9.1. The Endocannabinoid System and Its Role in Brain Aging
9.2. The Endocannabinoidome
9.3. Endocannabinoidome and Neuroinflammation
10. Pro-Resolving Mediators in the Resolution of Age-Related Neuroinflammation
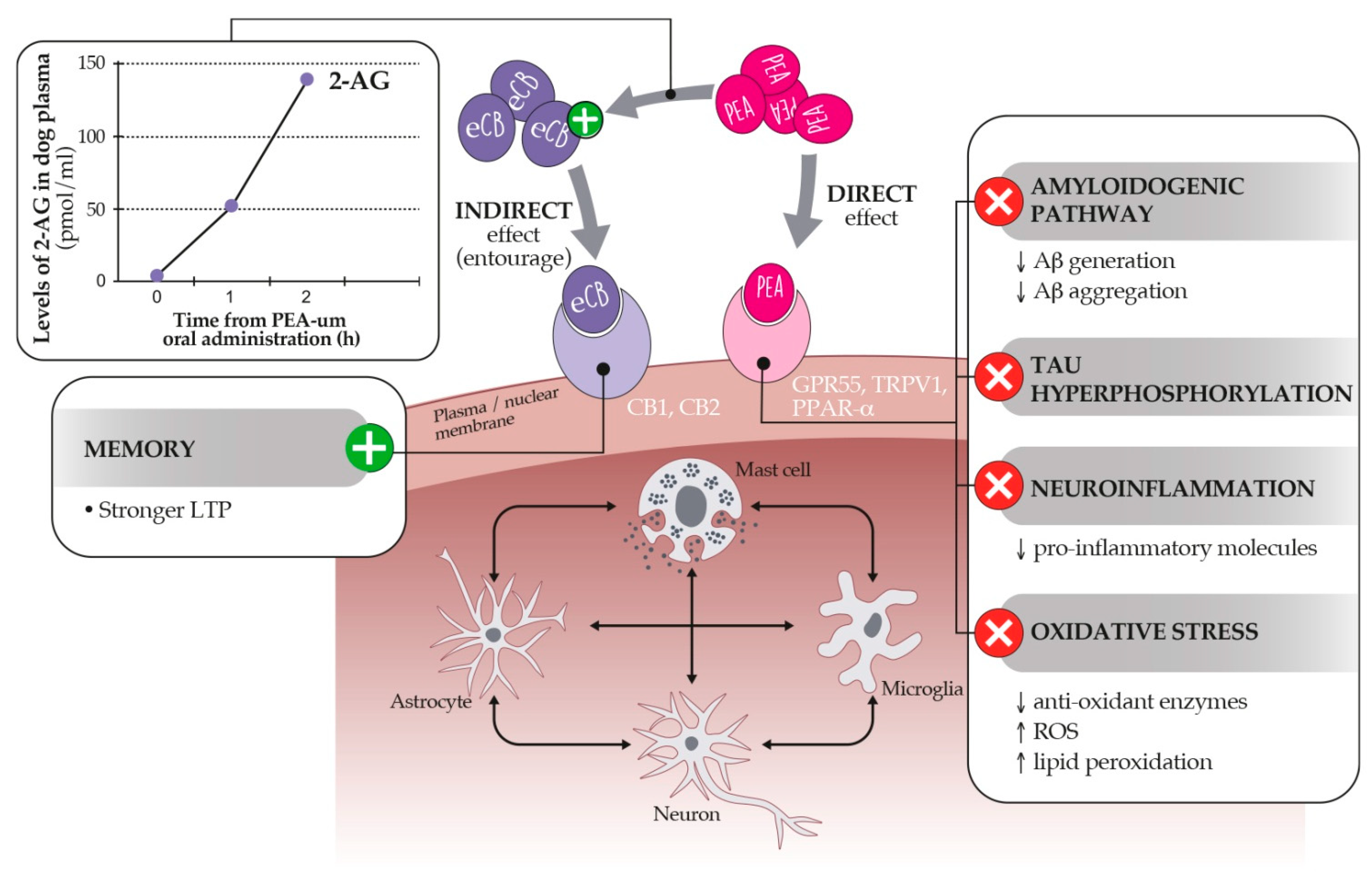
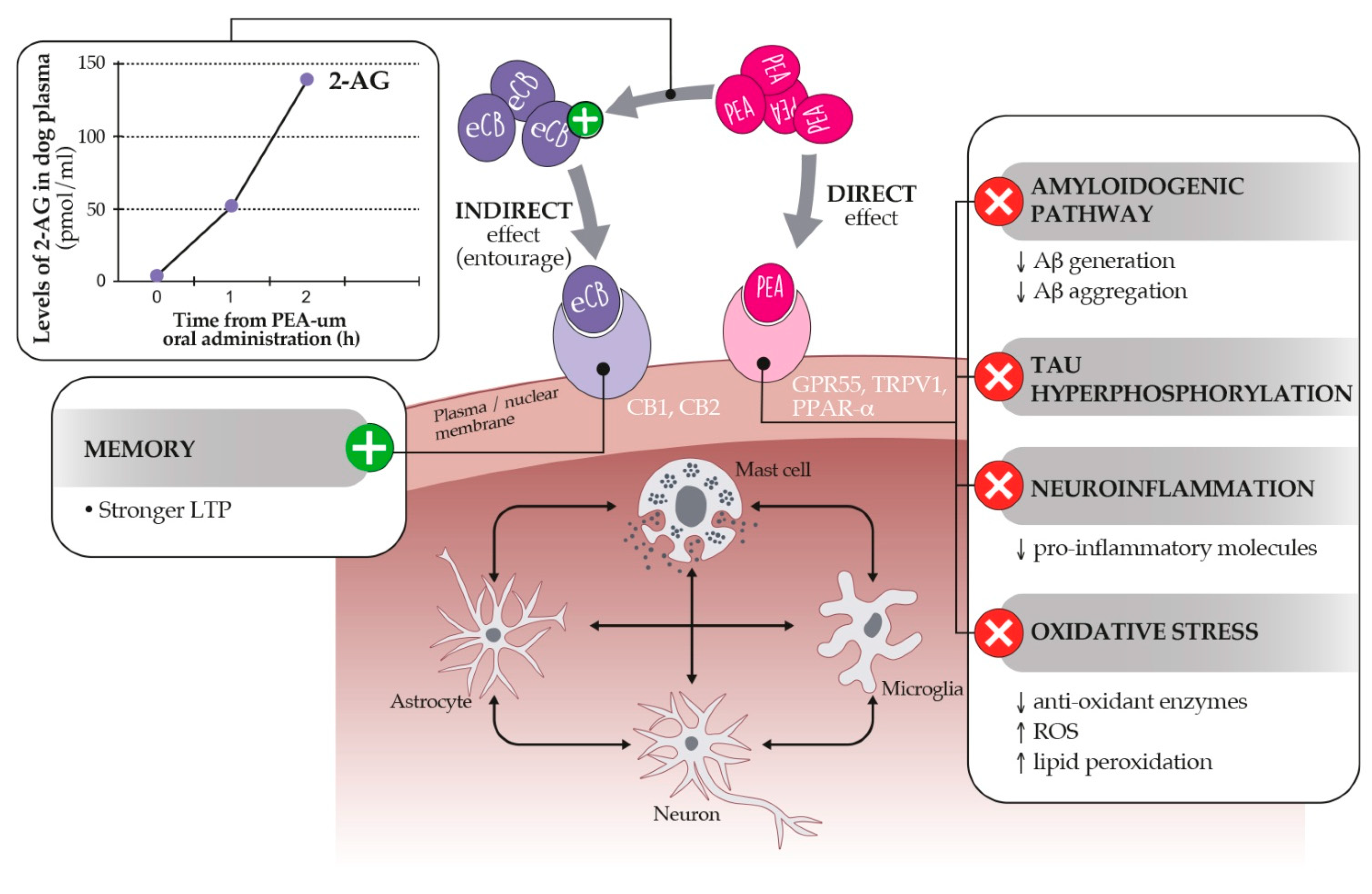
10.1. Palmitoylethanolamide: An Endocannabinoid Congener Endowed with Promising Anti-Inflammatory and Neuroprotective Properties
10.2. Dietary Supplementation with PEA-um as a Strategy to Control Age-Related Neuroinflammation and Neurobehavioral Correlates
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoylglycerol |
| AD | Alzheimer’s disease |
| AEA | anandamide |
| ALIA | Autacoid Local Injury Antagonism |
| BBB | blood-brain barrier |
| CA1 | cornu Ammonis 1 |
| CA3 | cornu Ammonis 3 |
| CB1 | cannabinoid receptor type 1 |
| CB2 | cannabinoid receptor type 2 |
| CCDRS | Canine Cognitive Dysfunction Rating Scale |
| CCDS | canine cognitive dysfunction syndrome |
| ccSDAT | Canine counterpart of senile dementia of Alzheimer’s type |
| CD33 | Cluster of differentiation 33 |
| CDS | cognitive dysfunction syndrome |
| CNS | central nervous system |
| COX-2 | cyclooxygenase-2 |
| D.I.S.H.A. | Disorientation, altered Interactions, Sleep-wake cycle changes, breaking in the House soiling and altered Activity levels |
| FAAH | fatty acid amide hydrolase |
| GPR18 | orphan G-protein coupled receptors 18 |
| GPR55 | orphan G-protein coupled receptors 55 |
| IL-1β | interleukin-1β |
| iNOS | inducible nitric oxide synthase |
| LTP | Long Term Potentiation |
| MCI | mild cognitive impairment |
| MRI | magnetic resonance imaging |
| PEA | palmitoylethanolamide |
| PPARα | peroxisome proliferator-activated receptor-α |
| PPARγ | peroxisome proliferator-activated receptor-γ |
| REM | rapid eye movement |
| TNFα | tumor necrosis factor α |
| TREM2 | triggering receptor expressed on myeloid cells 2 |
| TRPV1 | transient receptor potential vanilloid type 1 channel |
| um-PEA | ultramicronized palmitoylethanolamide |
References
- Scherbov, S.; Sanderson, W.C. New Approaches to the Conceptualization and Measurement of Age and Ageing. In Developments in Demographic Forecasting; The Springer Series on Demographic Methods and Population, Analysis; Mazzuco, S., Keilman, N., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 243–258. [Google Scholar] [CrossRef]
- Gunn-Moore, D. Considering older cats. J. Small Anim. Pract. 2006, 47, 430–431. [Google Scholar] [CrossRef]
- Quimby, J.; Gowland, S.; Carney, H.C.; DePorter, T.; Plummer, P.; Westropp, J. 2021 AAHA/AAFP Feline Life Stage Guidelines. J. Feline Med. Surg. 2021, 23, 211–233. [Google Scholar] [CrossRef]
- Creevy, K.E.; Grady, J.; Little, S.E.; Moore, G.E.; Strickler, B.G.; Thompson, S.; Webb, J.A. 2019 AAHA Canine Life Stage Guidelines. J. Am. Anim. Hosp. Assoc. 2019, 55, 267–290. [Google Scholar] [CrossRef]
- Urtamo, A.; Jyväkorpi, S.; Strandberg, T. Definitions of successful ageing: A brief review of a multidimensional concept. Acta Biomed. Atenei Parm. 2019, 90, 359–363. [Google Scholar] [CrossRef]
- McCune, S.; Stevenson, J.; Fretwell, L.; Thompson, A.; Mills, D.S. Ageing does not significantly affect performance in a spatial learning task in the domestic cat (felis silvestris catus). Appl. Anim. Behav. Sci. 2008, 112, 345–356. [Google Scholar] [CrossRef]
- Chapagain, D.; Range, F.; Huber, L.; Virányi, Z. Cognitive Aging in Dogs. Gerontology 2017, 64, 165–171. [Google Scholar] [CrossRef]
- Salvin, H.E.; McGreevy, P.D.; Sachdev, P.; Valenzuela, M. Growing old gracefully—Behavioral changes associated with “successful aging” in the dog, Canis familiaris. J. Vet. Behav. 2011, 6, 313–320. [Google Scholar] [CrossRef] [Green Version]
- González-Martínez, I.; Rosado, B.; Pesini, P.; García-Belenguer, S.; Palacio, J.; Villegas, A.; Suárez, M.-L.; Santamarina, G.; Sarasa, M. Effect of age and severity of cognitive dysfunction on two simple tasks in pet dogs. Vet. J. Lond. Engl. 1997, 198, 176–181. [Google Scholar] [CrossRef]
- Mongillo, P.; Pitteri, E.; Carnier, P.; Gabai, G.; Adamelli, S.; Marinelli, L. Does the attachment system towards owners change in aged dogs? Physiol. Behav. 2013, 120, 64–69. [Google Scholar] [CrossRef]
- Turcsán, B.; Wallis, L.; Berczik, J.; Range, F.; Kubinyi, E.; Virányi, Z. Individual and group level personality change across the lifespan in dogs. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Van Bourg, J.; Gilchrist, R.; Wynne, C.D.L. Adaptive spatial working memory assessments for aging pet dogs. Anim. Cogn. 2020, 24, 511–531. [Google Scholar] [CrossRef]
- Mongillo, P.; Araujo, J.A.; Pitteri, E.; Carnier, P.; Adamelli, S.; Regolin, L.; Marinelli, L. Spatial reversal learning is impaired by age in pet dogs. AGE Dordr. Neth. 2013, 35, 2273–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banzato, T.; Franzo, G.; Di Maggio, R.; Nicoletto, E.; Burti, S.; Cesari, M.; Canevelli, M. A Frailty Index based on clinical data to quantify mortality risk in dogs. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.; Carney, H.C.; Boynton, B.; Quimby, J.; Robertson, S.; Denis, K.S.; Tuzio, H.; Wright, B. 2021 AAFP Feline Senior Care Guidelines. J. Feline Med. Surg. 2021, 23, 613–638. [Google Scholar] [CrossRef]
- Head, E.; Mehta, R.; Hartley, J.; Kameka, M.; Cummings, B.J.; Cotman, C.W.; Ruehl, W.W.; Milgram, N.W. Spatial learning and memory as a function of age in the dog. Behav. Neurosci. 1995, 109, 851–858. [Google Scholar] [CrossRef]
- Chapagain, D.; Wallis, L.J.; Range, F.; Affenzeller, N.; Serra, J.; Virányi, Z. Behavioural and cognitive changes in aged pet dogs: No effects of an enriched diet and lifelong training. PLoS ONE 2020, 15, e0238517. [Google Scholar] [CrossRef] [PubMed]
- Golini, L.; Colangeli, R.; Tranquillo, V.; Mariscoli, M. Association between neurologic and cognitive dysfunction signs in a sample of aging dogs. J. Vet. Behav. 2009, 4, 25–30. [Google Scholar] [CrossRef]
- Youssef, S.A.; Capucchio, M.T.; Rofina, J.E.; Chambers, J.; Uchida, K.; Nakayama, H.; Head, E. Pathology of the Aging Brain in Domestic and Laboratory Animals, and Animal Models of Human Neurodegenerative Diseases. Vet. Pathol. 2016, 53, 327–348. [Google Scholar] [CrossRef]
- Russell, M.J.; Bobik, M.; White, R.G.; Hou, Y.; Benjamin, S.A.; Geddes, J.W. Age-specific onset of β-amyloid in Beagle brains. Neurobiol. Aging 1996, 17, 269–273. [Google Scholar] [CrossRef]
- Cummings, B.J.; Satou, T.; Head, E.; Milgram, N.W.; Cole, G.M.; Savage, M.J.; Podlisny, M.B.; Selkoe, D.J.; Siman, R.; Greenberg, B.D.; et al. Diffuse plaques contain C-terminal Aβ42 and not Aβ40: Evidence from cats and dogs. Neurobiol. Aging 1996, 17, 653–659. [Google Scholar] [CrossRef]
- Landsberg, G.M.; Nichol, J.; Araujo, J.A. Cognitive Dysfunction Syndrome: A Disease of Canine and Feline Brain Aging. Vet. Clin. N. Am. Small Anim. Pract. 2012, 42, 749–768. [Google Scholar] [CrossRef]
- Rofina, J.; van Ederen, A.; Toussaint, M.; Secrève, M.; van der Spek, A.; van der Meer, I.; Van Eerdenburg, F.; Gruys, E. Cognitive disturbances in old dogs suffering from the canine counterpart of Alzheimer’s disease. Brain Res. 2006, 1069, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Madari, A.; Farbakova, J.; Katina, S.; Smolek, T.; Novak, P.; Weissova, T.; Novak, M.; Zilka, N. Assessment of severity and progression of canine cognitive dysfunction syndrome using the CAnine DEmentia Scale (CADES). Appl. Anim. Behav. Sci. 2015, 171, 138–145. [Google Scholar] [CrossRef]
- Pageat, P.; Zecchini, M.; Verga, M.; Carenzi, C. Patologia Comportamentale del Cane; Le Point Veterinaire Italie: Milano, Italia, 1999. [Google Scholar]
- Ruehl, W.; Bruyette, D.; DePaoli, A.; Cotman, C.; Head, E.; Milgram, N.; Cummings, B. Chapter 22 Canine cognitive dysfunction as a model for human age-related cognitive decline, dementia and Alzheimer’s disease: Clinical presentation, cognitive testing, pathology and response to 1-deprenyl therapy. Prog. Brain Res. 1995, 106, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Siwak, C.T.; Tapp, P.D.; Milgram, N.W. Effect of Age and Level of Cognitive Function on Spontaneous and Exploratory Behaviors in the Beagle Dog. Learn. Mem. 2001, 8, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fast, R.; Schütt, T.; Toft, N.; Møller, A.; Berendt, M. An Observational Study with Long-Term Follow-Up of Canine Cognitive Dysfunction: Clinical Characteristics, Survival, and Risk Factors. J. Vet. Intern. Med. 2013, 27, 822–829. [Google Scholar] [CrossRef]
- Ozawa, M.; Inoue, M.; Uchida, K.; Chambers, J.; Takeuch, Y.; Nakayama, H. Physical signs of canine cognitive dysfunction. J. Vet. Med. Sci. 2019, 81, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Salvin, H.E.; McGreevy, P.D.; Sachdev, P.S.; Valenzuela, M.J. The canine cognitive dysfunction rating scale (CCDR): A data-driven and ecologically relevant assessment tool. Vet. J. Lond. Engl. 1997, 188, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Vikartovska, Z.; Farbakova, J.; Smolek, T.; Hanes, J.; Zilka, N.; Hornakova, L.; Humenik, F.; Maloveska, M.; Hudakova, N.; Cizkova, D. Novel Diagnostic Tools for Identifying Cognitive Impairment in Dogs: Behavior, Biomarkers, and Pathology. Front. Vet. Sci. 2021, 7, 551895. [Google Scholar] [CrossRef]
- Gunn-Moore, D.; Moffat, K.; Christie, L.-A.; Head, E. Cognitive dysfunction and the neurobiology of ageing in cats. J. Small Anim. Pr. 2007, 48, 546–553. [Google Scholar] [CrossRef]
- Landsberg, G.M.; Malamed, R. Clinical Picture of Canine and Feline Cognitive Impairment. In Canine and Feline Dementia: Molecular Basis, Diagnostics and Therapy; Landsberg, G., Maďari, A., Žilka, N., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–12. [Google Scholar] [CrossRef]
- Takeuchi, T.; Harada, E. Age-related changes in sleep-wake rhythm in dog. Behav. Brain Res. 2002, 136, 193–199. [Google Scholar] [CrossRef]
- De Mendonça, A.; Ribeiro, F.; Guerreiro, M.; Palma, T.; Garcia, C.; Mendonca, A. Clinical significance of subcortical vascular disease in patients with mild cognitive impairment. Eur. J. Neurol. 2005, 12, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Urbanowitsch, N.; Degen, C.; Toro, P.; Schröder, J. Neurological Soft Signs in Aging, Mild Cognitive Impairment, and Alzheimer’s Disease—The Impact of Cognitive Decline and Cognitive Reserve. Front. Psychiatry 2015, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Boyle, P.A.; Wilson, R.S.; Aggarwal, N.T.; Arvanitakis, Z.; Kelly, J.; Bienias, J.L.; Bennett, D.A. Parkinsonian signs in subjects with mild cognitive impairment. Neurology 2005, 65, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.N.; Pugliese, M.; Gimeno-Bayón, J.; Rodríguez, M.J.; Mahy, N. Dogs with cognitive dysfunction syndrome: A natural model of Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 298–314. [Google Scholar] [CrossRef] [Green Version]
- Studzinski, C.M.; Araujo, J.A.; Milgram, N.W. The canine model of human cognitive aging and dementia: Pharmacological validity of the model for assessment of human cognitive-enhancing drugs. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2005, 29, 489–498. [Google Scholar] [CrossRef]
- Chambers, J.; Tokuda, T.; Uchida, K.; Ishii, R.; Tatebe, H.; Takahashi, E.; Tomiyama, T.; Une, Y.; Nakayama, H. The domestic cat as a natural animal model of Alzheimer’s disease. Acta Neuropathol. Commun. 2015, 3, 78. [Google Scholar] [CrossRef] [Green Version]
- Gołaszewska, A.; Bik, W.; Motyl, T.; Orzechowski, A. Bridging the Gap between Alzheimer’s Disease and Alzheimer’s-like Diseases in Animals. Int. J. Mol. Sci. 2019, 20, 1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, E.; Moffat, K.; Das, P.; Sarsoza, F.; Poon, W.; Landsberg, G.; Cotman, C.W.; Murphy, M. β-Amyloid deposition and tau phosphorylation in clinically characterized aged cats. Neurobiol. Aging 2005, 26, 749–763. [Google Scholar] [CrossRef]
- Landsberg, G.; Araujo, J.A. Behavior Problems in Geriatric Pets. Vet. Clin. N. Am. Small Anim. Pract. 2005, 35, 675–698. [Google Scholar] [CrossRef]
- Colangeli, R.; Fassola, F.; Furlanello, T.; Giussani, S.; Osella, M.C.; Petrantoni, G.; Severi, E.; Sgarbi, C. Riconoscere e monitorare i segni clinici di invecchiamento cerebrale nel cane: Una metodologia per il veterinario generalista. Veterinaria 2005, 19, 19–23. [Google Scholar]
- Colle, M.; Hauw, J.J.; Crespeau, F.; Uchihara, T.; Akiyama, H.; Checler, F.; Pageat, P.; Duykaerts, C. Vascular and parenchymal Abeta deposition in the aging dog: Correlation with behavior. Neurobiol. Aging 2000, 21, 695–704. [Google Scholar] [CrossRef]
- Pugliese, M.; Carrasco, J.L.; Andrade, C.; Mas, E.; Mascort, J.; Mahy, N. Severe cognitive impairment correlates with higher cerebrospinal fluid levels of lactate and pyruvate in a canine model of senile dementia. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2005, 29, 603–610. [Google Scholar] [CrossRef]
- Snyder, J.M.; Shofer, F.S.; Winkle, T.J.; Massicotte, C. Canine Intracranial Primary Neoplasia: 173 Cases (1986-2003). J. Vet. Intern. Med. 2006, 20, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.; Lipitz, L.; Skorupski, K.; Shofer, F.; Van Winkle, T. Secondary Intracranial Neoplasia in the Dog: 177 Cases (1986-2003). J. Vet. Intern. Med. 2008, 22, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Mertens, M.; Fossum, T.W.; Willard, M.D.; Fosgate, G.; De La Paz, A.G.; Farmer, R.; Miller, M.W. Diagnosis of Congenital Portosystemic Shunt in Miniature Schnauzers 7 Years of Age or Older (1997–2006). J. Am. Anim. Hosp. Assoc. 2010, 46, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.; Rishniw, M. Periodontal disease is associated with cognitive dysfunction in aging dogs: A blinded prospective comparison of visual periodontal and cognitive questionnaire scores. Open Vet. J. 2021, 11, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Borràs, D.; Ferrer, I.; Pumarola, M. Age-related changes in the brain of the dog. Veter Pathol. 1999, 36, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Piguet, O.; Double, K.; Kril, J.; Harasty, J.; Macdonald, V.; McRitchie, D.; Halliday, G. White matter loss in healthy ageing: A postmortem analysis. Neurobiol. Aging 2009, 30, 1288–1295. [Google Scholar] [CrossRef]
- Chambers, J.K.; Uchida, K.; Nakayama, H. White matter myelin loss in the brains of aged dogs. Exp. Gerontol. 2011, 47, 263–269. [Google Scholar] [CrossRef]
- Su, M.; Head, E.; Brooks, W.M.; Wang, Z.; Muggenburg, B.A.; Adam, G.E.; Sutherland, R.; Cotman, C.W.; Nalcioglu, O. Magnetic resonance imaging of anatomic and vascular characteristics in a canine model of human aging. Neurobiol. Aging 1998, 19, 479–485. [Google Scholar] [CrossRef]
- Barry, E.F.; Loftus, J.P.; Luh, W.-M.; de Leon, M.J.; Niogi, S.N.; Johnson, P.J. Diffusion tensor-based analysis of white matter in the healthy aging canine brain. Neurobiol. Aging 2021, 105, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, M.; Carrasco, J.L.; Gomez-Anson, B.; Andrade, C.; Zamora, A.; Rodriguez, M.J.; Mascort, J.; Mahy, N. Magnetic resonance imaging of cerebral involutional changes in dogs as markers of aging: An innovative tool adapted from a human visual rating scale. Vet. J. 2010, 186, 166–171. [Google Scholar] [CrossRef]
- Dewey, C.W.; Rishniw, M.; Johnson, P.J.; Platt, S.; Robinson, K.; Sackman, J.; O’Donnell, M. Canine cognitive dysfunction patients have reduced total hippocampal volume compared with aging control dogs: A comparative magnetic resonance imaging study. Open Vet. J. 2021, 10, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, D.; Yayoshi, N.; Fujita, Y.; Fujita, M.; Orima, H. Measurement of interthalamic adhesion thickness as a criteria for brain atrophy in dogs with and without cognitive dysfunction (dementia). Vet. Radiol. Ultrasound Off. J. Am. Coll. Vet. Radiol. Int. Vet. Radiol. Assoc. 2005, 46, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.W.; Rishniw, M.; Johnson, P.J.; Davies, E.S.; Sackman, J.J.; O’Donnell, M.; Platt, S.; Robinson, K. Interthalamic adhesion size in aging dogs with presumptive spontaneous brain microhemorrhages: A comparative retrospective MRI study of dogs with and without evidence of canine cognitive dysfunction. PeerJ 2020, 8, e9012. [Google Scholar] [CrossRef]
- Scarpante, E.; Cherubini, G.B.; Stefani, A.; Taeymans, O. Magnetic resonance imaging features of leukoaraiosis in elderly dogs. Vet. Radiol. Ultrasound Off. J. Am. Coll. Vet. Radiol. Int. Vet. Radiol. Assoc. 2017, 58, 389–398. [Google Scholar] [CrossRef]
- Stylianaki, I.; Polizopoulou, Z.S.; Theodoridis, A.; Koutouzidou, G.; Baka, R.; Papaioannou, N.G. Amyloid-beta plasma and cerebrospinal fluid biomarkers in aged dogs with cognitive dysfunction syndrome. J. Vet. Intern. Med. 2020, 34, 1532–1540. [Google Scholar] [CrossRef]
- González-Martínez, I.; Rosado, B.; Pesini, P.; Suárez, M.-L.; Santamarina, G.; García-Belenguer, S.; Villegas, A.; Monleón, I.; Sarasa, M. Plasma β-amyloid peptides in canine aging and cognitive dysfunction as a model of Alzheimer’s disease. Exp. Gerontol. 2011, 46, 590–596. [Google Scholar] [CrossRef] [Green Version]
- Schütt, T.; Toft, N.; Berendt, M. Cognitive Function, Progression of Age-related Behavioral Changes, Biomarkers, and Survival in Dogs More Than 8 Years Old. J. Vet. Intern. Med. 2015, 29, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Panek, W.K.; Murdoch, D.M.; Gruen, M.E.; Mowat, F.M.; Marek, R.D.; Olby, N.J. Plasma Amyloid Beta Concentrations in Aged and Cognitively Impaired Pet Dogs. Mol. Neurobiol. 2020, 58, 483–489. [Google Scholar] [CrossRef]
- Phochantachinda, S.; Chantong, B.; Reamtong, O.; Chatchaisak, D. Change in the plasma proteome associated with canine cognitive dysfunction syndrome (CCDS) in Thailand. BMC Vet. Res. 2021, 17, 1–14. [Google Scholar] [CrossRef]
- Bain, M.J.; Hart, B.L.; Cliff, K.D.; Ruehl, W.W. Predicting behavioral changes associated with age-related cognitive impairment in dogs. J. Am. Vet. Med. Assoc. 2001, 218, 1792–1795. [Google Scholar] [CrossRef] [PubMed]
- Neilson, J.C.; Hart, B.; Cliff, K.D.; Ruehl, W.W. Prevalence of behavioral changes associated with age-related cognitive impairment in dogs. J. Am. Vet. Med. Assoc. 2001, 218, 1787–1791. [Google Scholar] [CrossRef]
- Osella, M.C.; Re, G.; Odore, R.; Girardi, C.; Badino, P.; Barbero, R.; Bergamasco, L. Canine cognitive dysfunction syndrome: Prevalence, clinical signs and treatment with a neuroprotective nutraceutical. Appl. Anim. Behav. Sci. 2007, 105, 297–310. [Google Scholar] [CrossRef]
- Katina, S.; Farbakova, J.; Madari, A.; Novak, M.; Zilka, N. Risk factors for canine cognitive dysfunction syndrome in Slovakia. Acta Vet. Scand. 2015, 58, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvin, H.E.; McGreevy, P.D.; Sachdev, P.S.; Valenzuela, M.J. Under diagnosis of canine cognitive dysfunction: A cross-sectional survey of older companion dogs. Vet. J. 2010, 184, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Azkona, G.; García-Belenguer, S.; Chacón, G.; Rosado, B.; León, M.; Palacio, J. Prevalence and risk factors of behavioural changes associated with age-related cognitive impairment in geriatric dogs. J. Small Anim. Pr. 2009, 50, 87–91. [Google Scholar] [CrossRef]
- Landsberg, G. Therapeutic agents for the treatment of cognitive dysfunction syndrome in senior dogs. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2005, 29, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.W.; Davies, E.S.; Xie, H.; Wakshlag, J.J. Canine Cognitive Dysfunction: Pathophysiology, Diagnosis, and Treatment. Vet. Clin. N. Am. Small Anim. Pract. 2019, 49, 477–499. [Google Scholar] [CrossRef]
- Tynes, V.V.; Landsberg, G.M. Nutritional Management of Behavior and Brain Disorders in Dogs and Cats. Vet. Clin. N. Am. Small Anim. Prat. 2021, 51, 711–727. [Google Scholar] [CrossRef]
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and Cognitive Impairment in Alzheimer Disease: A Complex but Coherent Relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef]
- Abey, A.; Davies, D.; Goldsbury, C.; Buckland, M.; Valenzuela, M.; Duncan, T. Distribution of tau hyperphosphorylation in canine dementia resembles early Alzheimer’s disease and other tauopathies. Brain Pathol. 2020, 31, 144–162. [Google Scholar] [CrossRef]
- Cummings, B.; Head, E.; Afagh, A.J.; Milgram, N.W.; Cotman, C.W. β-Amyloid Accumulation Correlates with Cognitive Dysfunction in the Aged Canine. Neurobiol. Learn. Mem. 1996, 66, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Smolek, T.; Madari, A.; Farbakova, J.; Kandrac, O.; Jadhav, S.; Cente, M.; Brezovakova, V.; Novak, M.; Zilka, N. Tau hyperphosphorylation in synaptosomes and neuroinflammation are associated with canine cognitive impairment. J. Comp. Neurol. 2015, 524, 874–895. [Google Scholar] [CrossRef]
- Schmidt, F.; Boltze, J.; Jäger, C.; Hofmann, S.; Willems, N.; Seeger, J.; Härtig, W.; Stolzing, A. Detection and Quantification of β-Amyloid, Pyroglutamyl Aβ, and Tau in Aged Canines. J. Neuropathol. Exp. Neurol. 2015, 74, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Head, E.; McCleary, R.; Hahn, F.; Milgram, N.; Cotman, C. Region-specific age at onset of β-amyloid in dogs. Neurobiol. Aging 2000, 21, 89–96. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Demonstration of Amyloid Deposits and Neurofibrillary Changes in Whole Brain Sections. Brain Pathol. 1991, 1, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Satou, T.; Cummings, B.; Head, E.; Nielson, K.; Hahn, F.F.; Milgram, N.W.; Velazquez, P.; Cribbs, D.H.; Tenner, A.J.; Cotman, C.W. The progression of β-amyloid deposition in the frontal cortex of the aged canine. Brain Res. 1997, 774, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Torp, R.; Head, E.; Cotman, C.W. Ultrastructural analyses of β-amyloid in the aged dog brain: Neuronal β-amyloid is localized to the plasma membrane. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2000, 24, 801–810. [Google Scholar] [CrossRef]
- Schütt, T.; Helboe, L.; Pedersen, L.; Waldemar, G.; Berendt, M.; Pedersen, J.T. Dogs with Cognitive Dysfunction as a Spontaneous Model for Early Alzheimer’s Disease: A Translational Study of Neuropathological and Inflammatory Markers. J. Alzheimers Dis. 2016, 52, 433–449. [Google Scholar] [CrossRef]
- Ozawa, M.; Chambers, J.; Uchida, K.; Nakayama, H. The Relation between canine cognitive dysfunction and age-related brain lesions. J. Vet. Med. Sci. 2016, 78, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Urfer, S.R.; Darvas, M.; Czeibert, K.; Sándor, S.; Promislow, D.E.L.; Creevy, K.E.; Kubinyi, E.; Kaeberlein, M. Canine Cognitive Dysfunction (CCD) Scores Correlate with Amyloid Beta 42 Levels in Dog Brain Tissue. GeroSci. 2021. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Castillo, C.; Hernández, J. The Role of Oxidative Stress in the Development of Cognitive Dysfunction Syndrome in Cats. Importance of Antioxidant Prevention and Therapy. SOJ Vet. Sci. 2015, 1, 1–12. [Google Scholar] [CrossRef]
- Head, E.; Thornton, P.L.; Tong, L.; Cotman, C.W. Initiation and propagation of molecular cascades in human brain aging: Insight from the canine model to promote successful aging. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2000, 24, 777–786. [Google Scholar] [CrossRef]
- Rusbridge, C.; Salguero, F.J.; David, M.A.; Faller, K.M.; Bras, J.T.; Guerreiro, R.; Richard-Londt, A.C.; Grainger, D.; Head, E.; Brandner, S.G.P.; et al. An Aged Canid with Behavioral Deficits Exhibits Blood and Cerebrospinal Fluid Amyloid Beta Oligomers. Front. Aging Neurosci. 2018, 10, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, P.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Onyango, I.; Jauregui, G.; Čarná, M.; Bennett, J.; Stokin, G. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Kempuraj, D.; Ahmed, M.E.; Selvakumar, G.P.; Thangavel, R.; Dhaliwal, A.S.; Dubova, I.; Mentor, S.; Premkumar, K.; Saeed, D.; Zahoor, H.; et al. Brain Injury–Mediated Neuroinflammatory Response and Alzheimer’s Disease. Neuroscience 2019, 26, 134–155. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, J.K.; Kulka, M. Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1093. [Google Scholar] [CrossRef] [PubMed]
- Verkhratskiĭ, A.N.; Butt, A. Glial Physiology and Pathophysiology; Wiley-Blackwell: Chichester, UK; Hoboken, NJ, USA, 2013. [Google Scholar]
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why are Astrocytes Important? Neurochem. Res. 2014, 40, 389–401. [Google Scholar] [CrossRef]
- Parpura, V.; Verkhratsky, A. Neuroglia at the Crossroads of Homoeostasis, Metabolism and Signalling: Evolution of the Concept. ASN Neuro 2012, 4, AN20120019. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Verkhratsky, A. The role of neuroglia in autism spectrum disorders. Prog. Mol. Biol. Transl. Sci. 2020, 173, 301–330. [Google Scholar] [CrossRef] [PubMed]
- Cartocci, V.; Catallo, M.; Tempestilli, M.; Segatto, M.; Pfrieger, F.; Bronzuoli, M.R.; Scuderi, C.; Servadio, M.; Trezza, V.; Pallottini, V. Altered Brain Cholesterol/Isoprenoid Metabolism in a Rat Model of Autism Spectrum Disorders. Neuroscience 2018, 372, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.; Zorec, R. Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Nayak, D.; Roth, T.; McGavern, D.B. Microglia Development and Function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [Green Version]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the Central Nervous System. Cell. Mol. Neurobiol. 2017, 38, 53–71. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, P. Mast Cells in Neuroimmune Interactions. Trends Neurosci. 2019, 42, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Facci, L.; Giusti, P. Neuroinflammation, Microglia and Mast Cells in the Pathophysiology of Neurocognitive Disorders: A Review. CNS Neurol. Disord. Drug Targets 2015, 13, 1654–1666. [Google Scholar] [CrossRef]
- Jones, M.K.; Nair, A.; Gupta, M. Mast Cells in Neurodegenerative Disease. Front. Cell. Neurosci. 2019, 13, 171. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Dong, H.; Xu, Y.; Zhang, S. Induction of Microglial Activation by Mediators Released from Mast Cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 38, 1520–1531. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, X.; Wang, Y.; Zhou, X.; Qian, Y.; Zhang, S. Suppression of Brain Mast Cells Degranulation Inhibits Microglial Activation and Central Nervous System Inflammation. Mol. Neurobiol. 2016, 54, 997–1007. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Giusti, P. Mast cells, glia and neuroinflammation: Partners in crime? Immunology 2014, 141, 314–327. [Google Scholar] [CrossRef]
- Mattila, O.S.; Strbian, D.; Saksi, J.; Pikkarainen, T.O.; Rantanen, V.; Tatlisumak, T.; Lindsberg, P.J. Cerebral Mast Cells Mediate Blood-Brain Barrier Disruption in Acute Experimental Ischemic Stroke Through Perivascular Gelatinase Activation. Stroke 2011, 42, 3600–3605. [Google Scholar] [CrossRef] [Green Version]
- Hendriksen, E.; van Bergeijk, D.; Oosting, R.S.; Redegeld, F.A. Mast cells in neuroinflammation and brain disorders. Neurosci. Biobehav. Rev. 2017, 79, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation—A common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sochocka, M.; Diniz, B.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2016, 54, 8071–8089. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Gilliam, E.A.; Li, L. Innate Immune Programing by Endotoxin and Its Pathological Consequences. Front. Immunol. 2015, 5, 680. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Fullerton, J.; Gilroy, D. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Steardo, L.; Bronzuoli, M.R.; Eiacomino, A.; Eesposito, G.; Escuderi, C. Does neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytes. Front. Neurosci. 2015, 9, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, E.M.; Initiative, T.A.D.N.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Rozemuller, A.J.M.; Van Haastert, E.S.; Eikelenboom, P.; Van Gool, W. Neuroinflammation in Alzheimer’s disease wanes with age. J. Neuroinflammation 2011, 8, 171. [Google Scholar] [CrossRef] [Green Version]
- King, E.; O’Brien, J.; Donaghy, P.; Morris, C.; Barnett, N.; Olsen, K.; Martin-Ruiz, C.; Taylor, J.-P.; Thomas, A.J. Peripheral inflammation in prodromal Alzheimer’s and Lewy body dementias. J. Neurol. Neurosurg. Psychiatry 2017, 89, 339–345. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting neuroinflammation in Alzheimer’s disease. J. Inflamm. Res. 2016, 9, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenza, M.; Facchinetti, R.; Steardo, L.; Scuderi, C. Altered Waste Disposal System in Aging and Alzheimer’s Disease: Focus on Astrocytic Aquaporin-4. Front. Pharmacol. 2020, 10, 1656. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L.; Scuderi, C. Astrocyte: An Innovative Approach for Alzheimer’s Disease Therapy. Curr. Pharm. Des. 2018, 23, 4979–4989. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Stecca, C.; Iacomino, A.; Steardo, L. Role of astrocytes in major neurological disorders: The evidence and implications. IUBMB Life 2013, 65, 957–961. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2004, 57, 67–81. [Google Scholar] [CrossRef]
- Morgan, J.T.; Chana, G.; Abramson, I.; Semendeferi, K.; Courchesne, E.; Everall, I.P. Abnormal microglial–neuronal spatial organization in the dorsolateral prefrontal cortex in autism. Brain Res. 2012, 1456, 72–81. [Google Scholar] [CrossRef]
- Tetreault, N.A.; Hakeem, A.Y.; Jiang, S.; Williams, B.A.; Allman, E.; Wold, B.J.; Allman, J.M. Microglia in the Cerebral Cortex in Autism. J. Autism Dev. Disord. 2012, 42, 2569–2584. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.; et al. Microglial Activation in Young Adults With Autism Spectrum Disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef]
- Thomsen, B.B.; Madsen, C.; Krohn, K.T.; Thygesen, C.; Schütt, T.; Metaxas, A.; Darvesh, S.; Agerholm, J.S.; Wirenfeldt, M.; Berendt, M.; et al. Mild Microglial Responses in the Cortex and Perivascular Macrophage Infiltration in Subcortical White Matter in Dogs with Age-Related Dementia Modelling Prodromal Alzheimer’s Disease. J. Alzheimers Dis. 2021, 82, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Paresce, D.M.; Ghosh, R.N.; Maxfield, F.R. Microglial Cells Internalize Aggregates of the Alzheimer’s Disease Amyloid β-Protein Via a Scavenger Receptor. Neuron 1996, 17, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Kummer, M.P.; Hammerschmidt, T.; Martinez, A.; Terwel, D.; Eichele, G.; Witten, A.; Figura, S.; Stoll, M.; Schwartz, S.; Pape, H.-C.; et al. Ear2 Deletion Causes Early Memory and Learning Deficits in APP/PS1 Mice. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 8845–8854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuderi, C.; Bronzuoli, M.R.; Facchinetti, R.; Pace, L.; Ferraro, L.; Broad, K.D.; Serviddio, G.; Bellanti, F.; Palombelli, G.; Carpinelli, G.; et al. Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer’s disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry 2018, 8, 32. [Google Scholar] [CrossRef]
- Olabarria, M.; Noristani, H.; Verkhratsky, A.; Rodríguez, J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef]
- Olabarria, M.; Noristani, H.; Verkhratsky, A.; Rodríguez, J.J. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: Mechanism for deficient glutamatergic transmission? Mol. Neurodegener. 2011, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.-Y.; Vadhwana, B.; Verkhratsky, A.; Rodriguez, J.J. Early Astrocytic Atrophy in the Entorhinal Cortex of a Triple Transgenic Animal Model of Alzheimer’s Disease. ASN Neuro 2011, 3, 271–279. [Google Scholar] [CrossRef]
- Kulijewicz-Nawrot, M.; Verkhratsky, A.; Chvatal, A.; Syková, E.; Rodríguez, J.J. Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J. Anat. 2012, 221, 252–262. [Google Scholar] [CrossRef]
- Beauquis, J.; Pavía, P.; Pomilio, C.; Vinuesa, A.; Podlutskaya, N.; Galvan, V.; Saravia, F. Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease. Exp. Neurol. 2013, 239, 28–37. [Google Scholar] [CrossRef]
- Gupta, K.; Harvima, I.T. Mast cell-neural interactions contribute to pain and itch. Immunol. Rev. 2018, 282, 168–187. [Google Scholar] [CrossRef]
- Harcha, P.A.; Vargas, A.; Yi, C.; Koulakoff, A.A.; Giaume, C.; Sáez, J.C. Hemichannels Are Required for Amyloid -Peptide-Induced Degranulation and Are Activated in Brain Mast Cells of APPswe/PS1dE9 Mice. J. Neurosci. 2015, 35, 9526–9538. [Google Scholar] [CrossRef] [Green Version]
- Shaik-Dasthagirisaheb, Y.B.; Conti, P. The Role of Mast Cells in Alzheimer’s Disease. Adv. Clin. Exp. Med. Off. Organ. Wroc. Med. Univ. 2016, 25, 781–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslinska, D.; Laure-Kamionowska, M.; Maslinski, K.T.; Gujski, M.; Maslinski, S. Distribution of tryptase-containing mast cells and metallothionein reactive astrocytes in human brains with amyloid deposits. Inflamm. Res. 2007, 56, S17–S18. [Google Scholar] [CrossRef] [PubMed]
- Harcha, P.A.; Garcés, P.; Arredondo, C.; Fernández, G.; Sáez, J.C.; van Zundert, B. Mast Cell and Astrocyte Hemichannels and Their Role in Alzheimer’s Disease, ALS, and Harmful Stress Conditions. Int. J. Mol. Sci. 2021, 22, 1924. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wang, Y.; Zhang, X.; Zhang, X.; Qian, Y.; Ding, H.; Zhang, S. Stabilization of Brain Mast Cells Alleviates LPS-Induced Neuroinflammation by Inhibiting Microglia Activation. Front. Cell. Neurosci. 2019, 13, 191. [Google Scholar] [CrossRef]
- Ruiz-Valdepeñas, L.; Benito, C.; Tolón, R.M.; Orgado, J.A.M.; Romero, J. The endocannabinoid system and amyloid-related diseases. Exp. Neurol. 2010, 224, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.; Hanus, L.; Breuer, A.; Pertwee, R.; Stevenson, L.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylgylcerol: A Possible Endogenous Cannabinoid Receptor Ligand in Brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.-C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Ohno-Shosaku, T.; Kano, M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr. Opin. Neurobiol. 2014, 29, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; Stella, N.; Zimmer, A. Endocannabinoid signalling and the deteriorating brain. Nat. Rev. Neurosci. 2014, 16, 30–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freundt-Revilla, J.; Heinrich, F.; Zoerner, A.; Gesell, F.; Beyerbach, M.; Shamir, M.; Oevermann, A.; Baumgärtner, W.; Tipold, A. The endocannabinoid system in canine Steroid-Responsive Meningitis-Arteritis and Intraspinal Spirocercosis. PLoS ONE 2018, 13, e0187197. [Google Scholar] [CrossRef] [Green Version]
- De Fonseca, F.R.; Ramos, J.A.; Bonnin, A.; Fernández-Ruiz, J.J. Presence of cannabinoid binding sites in the brain from early postnatal ages. Neuroreport 1993, 4, 135–138. [Google Scholar] [CrossRef]
- Glass, M.; Faull, R.; Dragunow, M. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318. [Google Scholar] [CrossRef]
- Freundt-Revilla, J.; Kegler, K.; Baumgärtner, W.; Tipold, A. Spatial distribution of cannabinoid receptor type 1 (CB1) in normal canine central and peripheral nervous system. PLoS ONE 2017, 12, e0181064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, R.J. The Endocannabinoid System of Animals. Animals 2019, 9, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilkei-Gorzo, A.; Drews, E.; Albayram, O.; Piyanova, A.; Gaffal, E.; Tueting, T.; Michel, K.; Mauer, D.; Maier, W.; Zimmer, A. Early onset of aging-like changes is restricted to cognitive abilities and skin structure in Cnr1−/− mice. Neurobiol. Aging 2010, 33, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Bab, I.; Zimmer, A. Cannabinoid receptors and the regulation of bone mass. Br. J. Pharmacol. 2008, 153, 182–188. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [Green Version]
- Taschler, U.; Radner, F.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Koefeler, H.C.; et al. Monoglyceride Lipase Deficiency in Mice Impairs Lipolysis and Attenuates Diet-induced Insulin Resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef] [Green Version]
- Morgese, M.G.; Cassano, T.; Cuomo, V.; Giuffrida, A. Anti-dyskinetic effects of cannabinoids in a rat model of Parkinson’s disease: Role of CB1 and TRPV1 receptors. Exp. Neurol. 2007, 208, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Scuderi, C.; Stecca, C.; Valenza, M.; Ratano, P.; Bronzuoli, M.R.; Bartoli, S.; Pompili, E.; Fumagalli, L.; Campolongo, P.; Steardo, L. Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1419-e1419. [Google Scholar] [CrossRef] [Green Version]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2019, 16, 9–29. [Google Scholar] [CrossRef]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of Endogenous Cannabinoids in Synaptic Signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [CrossRef]
- Carrera, J.; Tomberlin, J.; Kurtz, J.; Karakaya, E.; Bostanciklioglu, M.; Albayram, O. Endocannabinoid Signaling for GABAergic-Microglia (Mis)Communication in the Brain Aging. Front. Neurosci. 2021, 14, 606808. [Google Scholar] [CrossRef] [PubMed]
- Kasatkina, L.; Rittchen, S.; Sturm, E. Neuroprotective and Immunomodulatory Action of the Endocannabinoid System under Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 5431. [Google Scholar] [CrossRef] [PubMed]
- Cabral, G.A.; Harmon, K.N.; Carlisle, S.J. Cannabinoid-mediated Inhibition of Inducible Nitric Oxide Production by Rat Microglial Cells: Evidence for CB1 Receptor Participation. Adv. Exp. Med. Biol. 2001, 493, 207–214. [Google Scholar] [CrossRef]
- Facchinetti, F.; Del Giudice, E.; Furegato, S.; Passarotto, M.; Leon, A. Cannabinoids ablate release of TNFalpha in rat microglial cells stimulated with lypopolysaccharide. Glia 2002, 41, 161–168. [Google Scholar] [CrossRef]
- Scuderi, C.; Valenza, M.; Stecca, C.; Esposito, G.; Carratù, M.R.; Steardo, L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-α. J. Neuroinflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Scuderi, C.; Steardo, L. Neuroglial Roots of Neurodegenerative Diseases: Therapeutic Potential of Palmitoylethanolamide in Models of Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2013, 12, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo, L., Jr.; Carnuccio, R.; De Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by β-amyloid peptide. J. Cell. Mol. Med. 2011, 15, 2664–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.; Iuvone, T.; Steardo, L. Cannabidiol Reduces Aβ-Induced Neuroinflammation and Promotes Hippocampal Neurogenesis through PPARγ Involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef]
- Esposito, G.; Iuvone, T.; Savani, C.; Scuderi, C.; De Filippis, D.; Papa, M.; Di Marzo, V.; Steardo, L. Opposing Control of Cannabinoid Receptor Stimulation on Amyloid-β-Induced Reactive Gliosis: In Vitro and in Vivo Evidence. J. Pharmacol. Exp. Ther. 2007, 322, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L.; De Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V. Cannabidiol in vivo blunts β-amyloid induced neuroinflammation by suppressing IL-1β and iNOS expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Cipriano, M.; Esposito, G.; Negro, L.; Capoccia, E.; Sarnelli, G.; Scuderi, C.; De Filippis, D.; Steardo, L.; Iuvone, T. Palmitoylethanolamide Regulates Production of Pro-Angiogenic Mediators in a Model of β Amyloid-Induced Astrogliosis In Vitro. CNS Neurol. Disord. Drug Targets 2015, 14, 828–837. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L.; Romano, A.; Stecca, C.; Passarella, S.; Cassano, T.; Scuderi, C. Palmitoylethanolamide Dampens Reactive Astrogliosis and Improves Neuronal Trophic Support in a Triple Transgenic Model of Alzheimer’s Disease: In Vitro and In Vivo Evidence. Oxidative Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Van Der Stelt, M.; Mazzola, C.; Esposito, G.; Matias, I.; Petrosino, S.; De Filippis, D.; Micale, V.; Steardo, L.; Drago, F.; Iuvone, T.; et al. Endocannabinoids and β-amyloid-induced neurotoxicity in vivo: Effect of pharmacological elevation of endocannabinoid levels. Cell. Mol. Life Sci. 2006, 63, 1410–1424. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol Promotes Amyloid Precursor Protein Ubiquitination and Reduction of Beta Amyloid Expression in SHSY5Y APP+ Cells Through PPARγ Involvement. Phytother. Res. 2013, 28, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Capoccia, E.; Cirillo, C.; Marchetto, A.; Tiberi, S.; Sawikr, Y.; Pesce, M.; D’Alessandro, A.; Scuderi, C.; Sarnelli, G.; Cuomo, R.; et al. S100B-p53 disengagement by pentamidine promotes apoptosis and inhibits cellular migration via aquaporin-4 and metalloproteinase-2 inhibition in C6 glioma cells. Oncol. Lett. 2015, 9, 2864–2870. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, M.; Sanchez, C.; Galve-Roperh, I. Control of the cell survival/death decision by cannabinoids. J. Mol. Med. 2000, 78, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Panikashvili, D.; Shohami, E. Cannabinoids and brain injury: Therapeutic implications. Trends Mol. Med. 2002, 8, 58–61. [Google Scholar] [CrossRef]
- Van Der Stelt, M.; Veldhuis, W.B.; Van Haaften, G.W.; Fezza, F.; Bisogno, T.; Bär, P.R.; Veldink, G.A.; Vliegenthart, J.F.G.; Di Marzo, V.; Nicolay, K. Exogenous Anandamide Protects Rat Brain against Acute Neuronal InjuryIn Vivo. J. Neurosci. 2001, 21, 8765–8771. [Google Scholar] [CrossRef] [Green Version]
- Milton, N.G. Anandamide and noladin ether prevent neurotoxicity of the human amyloid-β peptide. Neurosci. Lett. 2002, 332, 127–130. [Google Scholar] [CrossRef]
- Parmentier-Batteur, S.; Jin, K.; Mao, X.O.; Xie, L.; Greenberg, D.A. Increased Severity of Stroke in CB1 Cannabinoid Receptor Knock-Out Mice. J. Neurosci. 2002, 22, 9771–9775. [Google Scholar] [CrossRef] [Green Version]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2Receptors and Fatty Acid Amide Hydrolase Are Selectively Overexpressed in Neuritic Plaque-Associated Glia in Alzheimer’s Disease Brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [Green Version]
- Bisogno, T. Cannabinoid Receptors and Endocannabinoids: Role in Neuroinflammatory and Neurodegenerative Disorders. CNS Neurol. Disord. Drug Targets 2010, 9, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive Lipids and Chronic Inflammation: Managing the Fire Within. Front. Immunol. 2018, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2015, 16, 51–67. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecha, M.; Carrillo-Salinas, F.J.; Feliu, A.; Mestre, L.; Guaza, C. Microglia activation states and cannabinoid system: Therapeutic implications. Pharmacol. Ther. 2016, 166, 40–55. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Barbierato, M.; Zusso, M.; Bruschetta, G.; Impellizzeri, D.; Cuzzocrea, S.; Giusti, P. N-Palmitoylethanolamine and Neuroinflammation: A Novel Therapeutic Strategy of Resolution. Mol. Neurobiol. 2015, 52, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Gokoh, M.; Kishimoto, S.; Oka, S.; Sugiura, T. 2-Arachidonoylglycerol Enhances the Phagocytosis of Opsonized Zymosan by HL-60 Cells Differentiated into Macrophage-Like Cells. Biol. Pharm. Bull. 2007, 30, 1199–1205. [Google Scholar] [CrossRef] [Green Version]
- Gugliandolo, E.; Peritore, A.F.; Piras, C.; Cuzzocrea, S.; Crupi, R. Palmitoylethanolamide and Related ALIAmides: Prohomeostatic Lipid Compounds for Animal Health and Wellbeing. Vet. Sci. 2020, 7, 78. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Giusti, P. Glia and Mast Cells as Targets for Palmitoylethanolamide, an Anti-inflammatory and Neuroprotective Lipid Mediator. Mol. Neurobiol. 2013, 48, 340–352. [Google Scholar] [CrossRef]
- Re, G.; Barbero, R.; Miolo, A.; Di Marzo, V.; Re, G.; Barbero, R.; Miolo, A.; Di Marzo, V. Palmitoylethanolamide, endocannabinoids and related cannabimimetic compounds in protection against tissue inflammation and pain: Potential use in companion animals. Vet. J. 2007, 173, 21–30. [Google Scholar] [CrossRef]
- Aloe, L.; Leon, A.; Levi-Montalcini, R. A proposed autacoid mechanism controlling mastocyte behaviour. Inflamm. Res. 1993, 39, C145–C147. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.; Vitale, R. The Endocannabinoid System and PPARs: Focus on Their Signalling Crosstalk, Action and Transcriptional Regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef] [PubMed]
- LoVerme, J.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The search for the palmitoylethanolamide receptor. Life Sci. 2005, 77, 1685–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.C.; Krebsbach, R.J.; Perry, S.R.; Dettmer, T.M.; Maasson, J.L.; Schmid, H.H. Occurrence and postmortem generation of anandamide and other long-chainN-acylethanolamines in mammalian brain. FEBS Lett. 1995, 375, 117–120. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.; Ortori, C.A.; Chapman, V.; Kendall, D.A.; Barrett, D.A. Quantitative profiling of endocannabinoids and related compounds in rat brain using liquid chromatography–tandem electrospray ionization mass spectrometry. Anal. Biochem. 2007, 360, 216–226. [Google Scholar] [CrossRef]
- Natarajan, V.; Schmid, P.C.; Reddy, P.V.; Zuzarte-Augustin, M.L.; Schmid, H.H.O. Biosynthesis of N-Acylethanolamine Phospholipids by Dog Brain Preparations. J. Neurochem. 1983, 41, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Franklin, A.; Witting, A.; Möller, T.; Stella, N. Astrocytes in Culture Produce Anandamide and Other Acylethanolamides. J. Biol. Chem. 2002, 277, 20869–20876. [Google Scholar] [CrossRef] [Green Version]
- Muccioli, G.; Stella, N. Microglia produce and hydrolyze palmitoylethanolamide. Neuropharmacology 2008, 54, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Bisogno, T.; Maurelli, S.; Melck, D.; De Petrocellis, L.; Di Marzo, V. Biosynthesis, Uptake, and Degradation of Anandamide and Palmitoylethanolamide in Leukocytes. J. Biol. Chem. 1997, 272, 3315–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenza, M.; Facchinetti, R.; Menegoni, G.; Steardo, L.; Scuderi, C. Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes. Biomolecules 2021, 11, 600. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Paterniti, I.; Mazzon, E.; Genovese, T.; di Paola, R.; Galuppo, M.; Cuzzocrea, S. Effects of palmitoylethanolamide on release of mast cell peptidases and neurotrophic factors after spinal cord injury. Brain Behav. Immun. 2011, 25, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Parrella, E.; Porrini, V.; Iorio, R.; Benarese, M.; Lanzillotta, A.; Mota, M.; Fusco, M.; Tonin, P.; Spano, P.; Pizzi, M. PEA and luteolin synergistically reduce mast cell-mediated toxicity and elicit neuroprotection in cell-based models of brain ischemia. Brain Res. 2016, 1648, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Petrosino, S.; Di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharmacol. 2016, 174, 1349–1365. [Google Scholar] [CrossRef]
- Alessio, N.; Belardo, C.; Trotta, M.; Paino, S.; Boccella, S.; Gargano, F.; Pieretti, G.; Ricciardi, F.; Marabese, I.; Luongo, L.; et al. Vitamin D Deficiency Induces Chronic Pain and Microglial Phenotypic Changes in Mice. Int. J. Mol. Sci. 2021, 22, 3604. [Google Scholar] [CrossRef]
- D’Aloia, A.; Molteni, L.; Gullo, F.; Bresciani, E.; Artusa, V.; Rizzi, L.; Ceriani, M.; Meanti, R.; Lecchi, M.; Coco, S.; et al. Palmitoylethanolamide Modulation of Microglia Activation: Characterization of Mechanisms of Action and Implication for Its Neuroprotective Effects: Palmitoylethanolamide and Its New Formulations. Int. J. Mol. Sci. 2021, 22, 3054. [Google Scholar] [CrossRef]
- Raso, G.M.; Russo, R.; Calignano, A.; Meli, R. Palmitoylethanolamide in CNS health and disease. Pharmacol. Res. 2014, 86, 32–41. [Google Scholar] [CrossRef]
- Petrosino, S.; Moriello, A.S.; Cerrato, S.; Fusco, M.; Puigdemont, A.; De Petrocellis, L.; Di Marzo, V. The anti-inflammatory mediator palmitoylethanolamide enhances the levels of 2-arachidonoyl-glycerol and potentiates its actions at TRPV1 cation channels. Br. J. Pharmacol. 2015, 173, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Piyanova, A.; Lomazzo, E.; Bindila, L.; Lerner, R.; Albayram, O.; Ruhl, T.; Lutz, B.; Zimmer, A.; Bilkei-Gorzo, A. Age-related changes in the endocannabinoid system in the mouse hippocampus. Mech. Ageing Dev. 2015, 150, 55–64. [Google Scholar] [CrossRef]
- Silva-Cruz, A.; Carlström, M.; Ribeiro, J.A.; Sebastião, A.M. Dual Influence of Endocannabinoids on Long-Term Potentiation of Synaptic Transmission. Front. Pharmacol. 2017, 8, 921. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, U.; Pelzer, M.; Kleine, J.; Hohmann, T.; Ghadban, C.; Dehghani, F. Opposite Effects of Neuroprotective Cannabinoids, Palmitoylethanolamide, and 2-Arachidonoylglycerol on Function and Morphology of Microglia. Front. Neurosci. 2019, 13, 1180. [Google Scholar] [CrossRef]
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Paterniti, I.; Cuzzocrea, S. Neuroprotective Activities of Palmitoylethanolamide in an Animal Model of Parkinson’s Disease. PLoS ONE 2012, 7, e41880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verme, J.L.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The Nuclear Receptor Peroxisome Proliferator-Activated Receptor-α Mediates the Anti-Inflammatory Actions of Palmitoylethanolamide. Mol. Pharmacol. 2004, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Borelli, A.C.; Ferraro, L.; Tanganelli, S.; Antonelli, T.; Tomasini, M.C. Palmitoylethanolamide Blunts Amyloid-β42-Induced Astrocyte Activation and Improves Neuronal Survival in Primary Mouse Cortical Astrocyte-Neuron Co-Cultures. J. Alzheimers Dis. 2017, 61, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Cassano, T.; Ferraro, L.; Tomasini, M.C. Astrocytic palmitoylethanolamide pre-exposure exerts neuroprotective effects in astrocyte-neuron co-cultures from a triple transgenic mouse model of Alzheimer’s disease. Life Sci. 2020, 257, 118037. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, S.; Strosznajder, J.B.; Jeżyna, M. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. GPR55: A new member of the cannabinoid receptor clan? Br. J. Pharmacol. 2007, 152, 984–986. [Google Scholar] [CrossRef] [Green Version]
- Zygmunt, P.M.; Ermund, A.; Movahed, P.; Andersson, D.; Simonsen, C.; Jönsson, B.; Blomgren, A.; Birnir, B.; Bevan, S.J.; Eschalier, A.; et al. Monoacylglycerols Activate TRPV1—A Link between Phospholipase C and TRPV1. PLoS ONE 2013, 8, e81618. [Google Scholar] [CrossRef] [Green Version]
- Bisogno, T. The Role of the Endocannabinoid System in Alzheimers Disease: Facts and Hypotheses. Curr. Pharm. Des. 2008, 14, 2299–2305. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, T.; Bartsch, J.C.; Beer, A.; Lomazzo, E.; Guggenhuber, S.; Lange, M.D.; Bindila, L.; Pape, H.-C.; Lutz, B. Impaired anandamide/palmitoylethanolamide signaling in hippocampal glutamatergic neurons alters synaptic plasticity, learning, and emotional responses. Neuropsychopharmacology 2018, 44, 1377–1388. [Google Scholar] [CrossRef]
- Petrosino, S.; Cordaro, M.; Verde, R.; Moriello, A.S.; Marcolongo, G.; Schievano, C.; Siracusa, R.; Piscitelli, F.; Peritore, A.F.; Crupi, R.; et al. Oral Ultramicronized Palmitoylethanolamide: Plasma and Tissue Levels and Spinal Anti-hyperalgesic Effect. Front. Pharmacol. 2018, 9, 249. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Crupi, R.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Micronized/ultramicronized palmitoylethanolamide displays superior oral efficacy compared to nonmicronized palmitoylethanolamide in a rat model of inflammatory pain. J. Neuroinflamm. 2014, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerrato, S.; Brazis, P.; della Valle, M.F.; Miolo, A.; Petrosino, S.; Di Marzo, V.; Puigdemont, A. Effects of palmitoylethanolamide on the cutaneous allergic inflammatory response in Ascaris hypersensitive Beagle dogs. Vet. J. 2012, 191, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Tomasini, M.C.; Cassano, T.; Ferraro, L. Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 428. [Google Scholar] [CrossRef] [Green Version]
- Caltagirone, C.; Stroke Study Group; Cisari, C.; Schievano, C.; di Paola, R.; Cordaro, M.; Bruschetta, G.; Esposito, E.; Cuzzocrea, S. Co-ultramicronized Palmitoylethanolamide/Luteolin in the Treatment of Cerebral Ischemia: From Rodent to Man. Transl. Stroke Res. 2015, 7, 54–69. [Google Scholar] [CrossRef] [Green Version]
- Cordaro, M.; Impellizzeri, D.; Paterniti, I.; Bruschetta, G.; Siracusa, R.; De Stefano, D.; Cuzzocrea, S.; Esposito, E. Neuroprotective Effects of Co-UltraPEALut on Secondary Inflammatory Process and Autophagy Involved in Traumatic Brain Injury. J. Neurotrauma 2016, 33, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Paterniti, I.; Ahmad, A.; Campolo, M.; Esposito, E.; Cuzzocrea, S. Effects of Palmitoylethanolamide and Luteolin in an Animal Model of Anxiety/Depression. CNS Neurol. Disord. Drug Targets 2013, 12, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a Potential Therapeutic Agent in Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 821. [Google Scholar] [CrossRef] [Green Version]
- Petrosino, S.; Moriello, A.S. Palmitoylethanolamide: A Nutritional Approach to Keep Neuroinflammation within Physiological Boundaries. A Systematic Review. Int. J. Mol. Sci. 2020, 21, 9526. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Cordaro, M.; Campolo, M.; Siracusa, R.; Cornelius, C.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Neuroprotection by Association of Palmitoylethanolamide with Luteolin in Experimental Alzheimer’s Disease Models: The Control of Neuroinflammation. CNS Neurol. Disord. Drug Targets 2014, 13, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, R.; Valenza, M.; Bronzuoli, M.R.; Menegoni, G.; Ratano, P.; Steardo, L.; Campolongo, P.; Scuderi, C. Looking for a Treatment for the Early Stage of Alzheimer’s Disease: Preclinical Evidence with Co-Ultramicronized Palmitoylethanolamide and Luteolin. Int. J. Mol. Sci. 2020, 21, 3802. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Bellanti, F.; Bukke, V.N.; Archana, M.; Scuderi, C.; Ferraro, L.; Serviddio, G.; Cuomo, V. Effects of Ultramicronized Palmitoylethanolamide Treatment on the Glutamatergic Alterations and Mitochondrial Impairment In 3 × Tg-Ad Mice. Pharmadvances 2021, 3, 87–88. [Google Scholar] [CrossRef]
- Assogna, M.; Casula, E.P.; Borghi, I.; Bonnì, S.; Samà, D.; Motta, C.; Di Lorenzo, F.; D’Acunto, A.; Porrazzini, F.; Minei, M.; et al. Effects of Palmitoylethanolamide Combined with Luteoline on Frontal Lobe Functions, High Frequency Oscillations, and GABAergic Transmission in Patients with Frontotemporal Dementia. J. Alzheimers Dis. 2020, 76, 1297–1308. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mental status and spatial orientation (confusional status) Get lost in a known environment Awaiting the door opening on the wrong side Inability to circumnavigate unknown objects Less interested in environmental stimuli |
| Relationships (social interaction) Less interested in being touched Ignoring the return of the owner Social behavior is disrupted Increased need for physical contact (is “needy”) |
| Activity (increased—repetitive) Starring at objects or empty space, fly biting Aimless walking Increased licking behavior (on the owner or objects) Increased vocalization |
| Activity (diminished) Apathetic, less interested in exploring Seems to not be interested anymore in known stimuli |
| Appetite Eats more than usual Eats less than usual |
| Toileting behavior Reduced time spent cleaning itself |
| Anxiety (irritability) Often irritable or anxious Shows signs of separation anxiety that has never had before Easily irritable |
| Sleep—awake cycle Short period of sleep interrupted by frequent abrupt awakenings Sleeps more than usual during daytime |
| Learning and memory Loss of housetraining, urinating or defecating in front of the owner Does not request to go out anymore Despite regular daily activity eliminates only when back home Eliminates where it sleepsIt is incontinent |
| Learned behavior and commands Struggle in performing a previously learned task Struggle to recognize a member of the family or other known people/animals Struggle to respond to commands Struggle to learn new commands or tasks |
 Astrocytes |
|
 Microglia |
|
 Mast cells |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scuderi, C.; Golini, L. Successful and Unsuccessful Brain Aging in Pets: Pathophysiological Mechanisms behind Clinical Signs and Potential Benefits from Palmitoylethanolamide Nutritional Intervention. Animals 2021, 11, 2584. https://doi.org/10.3390/ani11092584
Scuderi C, Golini L. Successful and Unsuccessful Brain Aging in Pets: Pathophysiological Mechanisms behind Clinical Signs and Potential Benefits from Palmitoylethanolamide Nutritional Intervention. Animals. 2021; 11(9):2584. https://doi.org/10.3390/ani11092584
Chicago/Turabian StyleScuderi, Caterina, and Lorenzo Golini. 2021. "Successful and Unsuccessful Brain Aging in Pets: Pathophysiological Mechanisms behind Clinical Signs and Potential Benefits from Palmitoylethanolamide Nutritional Intervention" Animals 11, no. 9: 2584. https://doi.org/10.3390/ani11092584
APA StyleScuderi, C., & Golini, L. (2021). Successful and Unsuccessful Brain Aging in Pets: Pathophysiological Mechanisms behind Clinical Signs and Potential Benefits from Palmitoylethanolamide Nutritional Intervention. Animals, 11(9), 2584. https://doi.org/10.3390/ani11092584