
Comparative Mitogenomics of True Frogs (Ranidae, Anura), and Its Implications for the Phylogeny and Evolutionary History of Rana


 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Library Preparation and Sequencing
2.3. Sequence Assembly and Annotation
2.4. Molecular Phylogenetic Analysis
2.5. Divergence Times Estimates
2.6. Mitogenomic Rearrangements Analysis
3. Results and Discussion
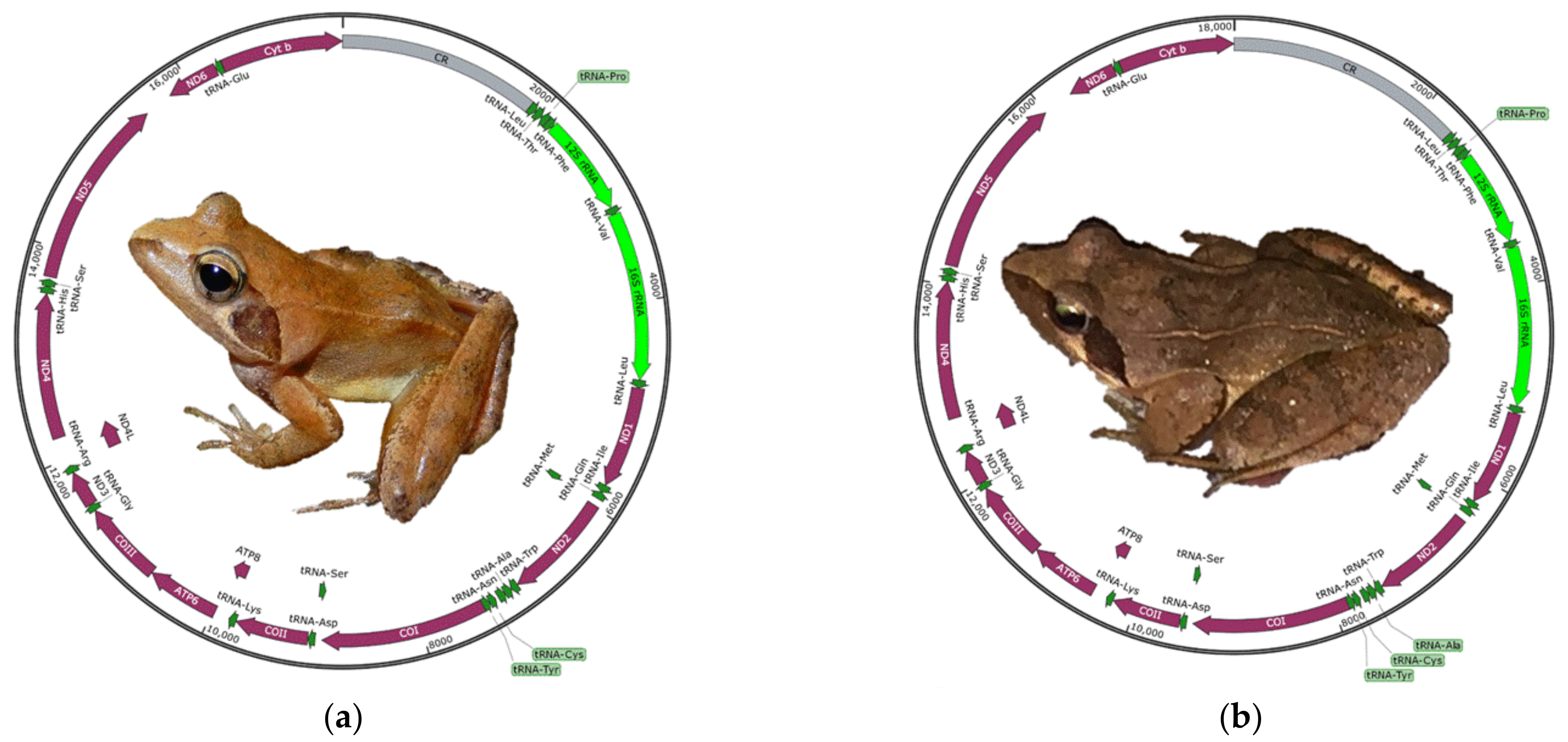
3.1. General Characteristics of the Mitogenome
3.2. Protein-Coding Genes and the Codon Usage
3.3. Transfer RNA, Ribosomal RNA Genes and Control Region
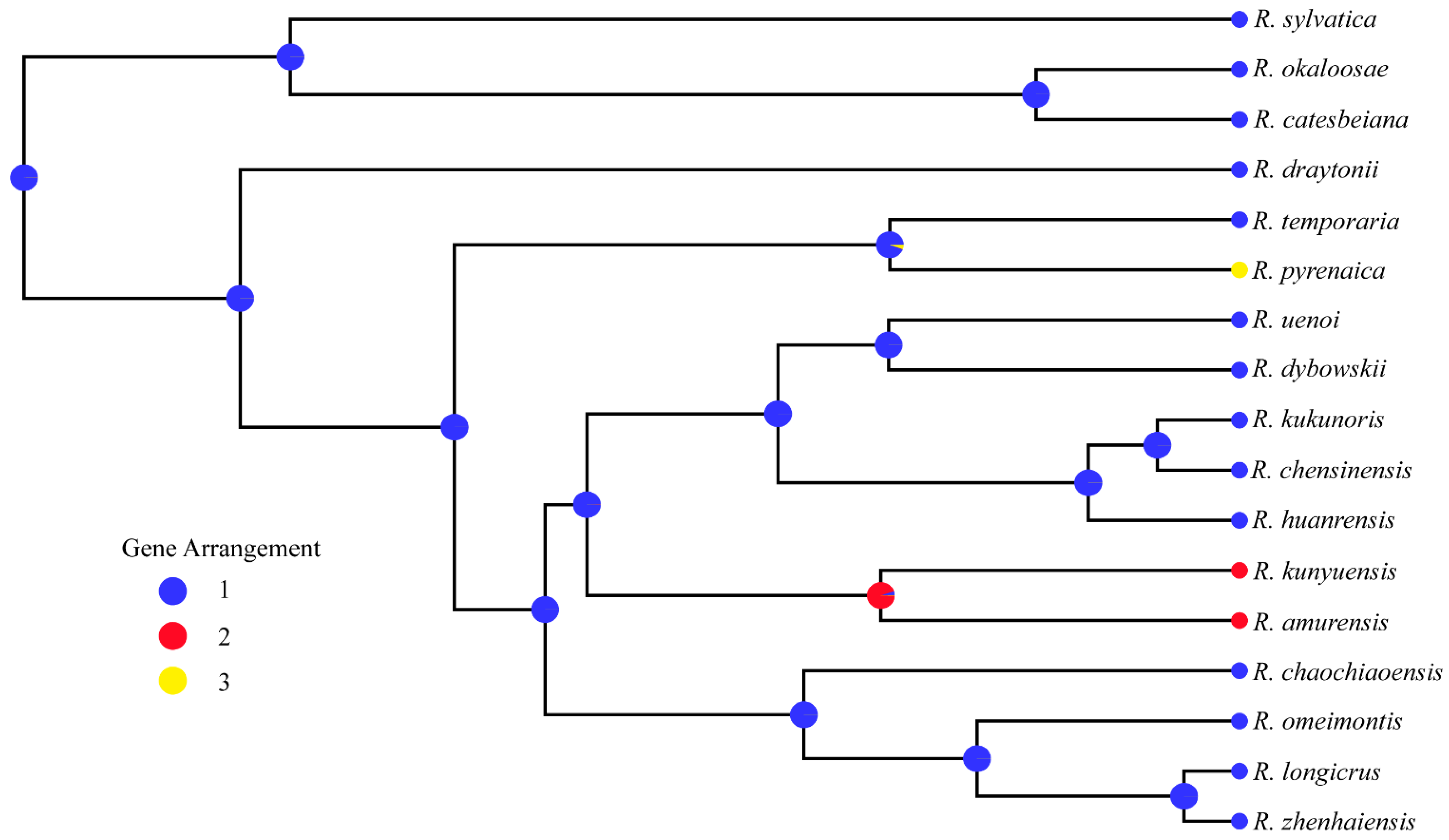
3.4. Phylogenetic Relationships
3.5. Divergence-Time Estimation
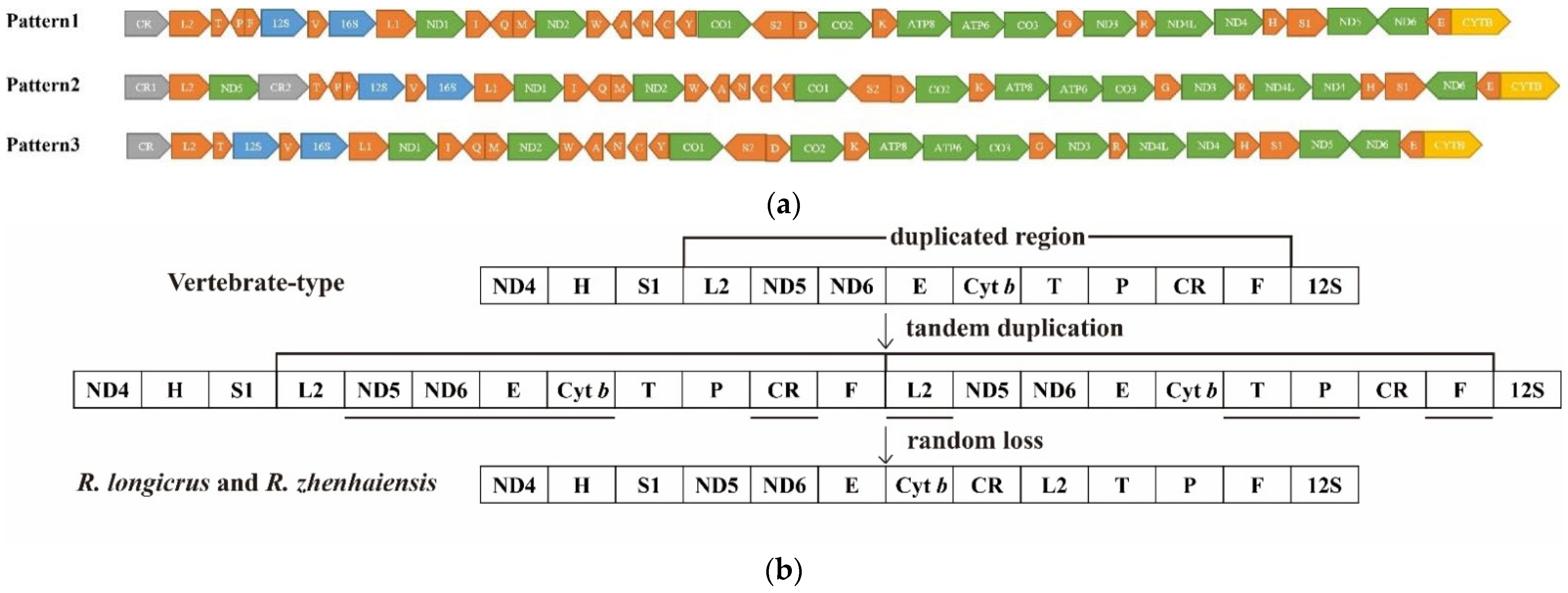
3.6. Mitogenomic Rearrangements
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frost, D.R. Amphibian Species of the World: An Online Reference, Version 6.0. 2019. Available online: http://research.amnh.org/vz/herpetology/amphibia (accessed on 24 October 2021).
- Yuan, Z.; Zhou, W.; Chen, X.; Poyarkov Jr, N.A.; Chen, H.; Jang-Liaw, N.; Chou, W.; Matzke, N.J.; Iizuka, K.; Min, M. Spatiotemporal diversification of the true frogs (genus Rana): A historical framework for a widely studied group of model organisms. Syst. Biol. 2016, 65, 824–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Zhou, K. Phylogenetic relationships among Chinese Ranids inferred from sequence data set of 12S and 16S rDNA. Herpetol. J. 2005, 15, 1–8. [Google Scholar]
- Wang, C.; Qian, L.; Zhang, C.; Guo, W.; Pan, T.; Wu, J.; Wang, H.; Zhang, B. A new species of Rana from the Dabie Mountains in eastern China (Anura, Ranidae). ZooKeys 2017, 724, 135–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiri, N.; Vaissi, S.; Aghamir, F.; Saberi-Pirooz, R.; Rödder, D.; Ebrahimi, E.; Ahmadzadeh, F. Tracking climate change in the spatial distribution pattern and the phylogeographic structure of Hyrcanian wood frog, Rana pseudodalmatina (Anura: Ranidae). J. Zool. Syst. Evol. Res. 2021, 59, 1604–1619. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Zhu, H.; Li, P.; Yang, B.; Ma, J. Phylogeny and biogeography of South Chinese brown frogs (Ranidae, Anura). PloS ONE 2017, 12, e0175113. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ren, J.; Chen, H.; Lyu, Z.; Guo, X.; Jiang, K.; Chen, J.; Li, J.; Guo, P.; Wang, Y. The updated checklists of amphibians and reptiles of China. Biodivers. Sci. 2020, 28, 189–218, (In Chinese with English Abstract). [Google Scholar]
- Che, J.; Pang, J.; Zhao, H.; Wu, G.; Zhao, E.; Zhang, Y. Phylogeny of Raninae (Anura: Ranidae) inferred from mitochondrial and nuclear sequences. Mol. Phylogenetics Evol. 2007, 43, 1–13. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, J.; Wang, C.; Li, P.; Murphy, R.W.; Che, J.; Yuan, Z. A new species of the genus Rana from Henan, central China (Anura, Ranidae). ZooKeys 2017, 694, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Lyu, Z.; Qi, S.; Zhao, J.; Li, P.; Wang, Y. A new species of the Rana japonica group (Anura, Ranidae, Rana) from China, with a taxonomic proposal for the R. johnsi group. ZooKeys 2020, 942, 141–158. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, S.; Zhang, H.; Chen, W.; Cai, B.; Hoang, V.; Wu, J.; Wang, B. A new species of the genus Rana sensu lato Linnaeus, 1758 (Anura, Ranidae) from Wuyi Mountain, Fujian Province, China. ZooKeys 2021, 1065, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhou, Y.; Min, M.; Matsui, M.; Dong, B.; Li, P.; Fong, J.J. Diversity and phylogeography of Northeast Asian brown frogs allied to Rana dybowskii (Anura, Ranidae). Mol. Phylogenet. Evol. 2017, 112, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yu, J.; Liu, J.; Zhou, M.; Li, B.; Ouyang, B. Three new Ranidae mitogenomes and the evolution of mitochondrial gene rearrangements among Ranidae species. Asian Herpetol. Res. 2018, 9, 85–98. [Google Scholar]
- Ruokonen, M.; Kvist, L. Structure and evolution of the avian mitochondrial control region. Mol. Phylogenet. Evol. 2002, 23, 422–432. [Google Scholar] [CrossRef]
- Oliver, L.A.; Prendini, E.; Kraus, F.; Raxworthy, C.J. Systematics and biogeography of the Hylarana frog (Anura: Ranidae) radiation across tropical Australasia, Southeast Asia, and Africa. Mol. Phylogenet. Evol. 2015, 90, 176–192. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Yoshimura, J.; Cai, W.; Sota, T.; Li, H. Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef]
- Hu, Y.; Thapa, A.; Fan, H.; Ma, T.; Wu, Q.; Ma, S.; Zhang, D.; Wang, B.; Li, M.; Yan, L. Genomic evidence for two phylogenetic species and long-term population bottlenecks in red pandas. Sci. Adv. 2020, 6, eaax5751. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wei, S.J.; Tang, P.; Wu, Q.; Shi, M.; Sharkey, M.J.; Chen, X.X. Multiple lines of evidence from mitochondrial genomes resolve phylogenetic relationships of parasitic wasps in Braconidae. Genome Biol. Evol. 2016, 8, 2651–2662. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; Sun, Z.; Lai, X.; Orozcoterwengel, P.; Yan, P.; Wu, G.; Wang, H.; Zhu, W.; Wu, X.; Zhang, B. Hidden species diversity in Pachyhynobius: A multiple approaches species delimitation with mitogenomes. Mol. Phylogenet. Evol. 2019, 137, 138–145. [Google Scholar] [CrossRef]
- Lamelas, L.; Aleix-Mata, G.; Rovatsos, M.; Marchal, J.A.; Palomeque, T.; Lorite, P.; Sánchez, A. Complete mitochondrial genome of three species of the genus Microtus (Arvicolinae, Rodentia). Animals 2020, 10, 2130. [Google Scholar] [CrossRef]
- Choi, E.; Kim, S.H.; Lee, S.J.; Jo, E.; Kim, J.; Kim, J.H.; Parker, S.J.; Chi, Y.M.; Park, H. A first genome survey and genomic SSR marker analysis of Trematomus loennbergii Regan, 1913. Animals 2021, 11, 3186. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Lobry, J.R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996, 13, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Z.; Gao, H.; Liu, Z.; Teng, L. The complete mitochondrial genome of the Rana kukunoris (Anura: Ranidae) from Inner Mongolia, China. Mitochondrial Dna Part B 2020, 5, 586–587. [Google Scholar] [CrossRef] [Green Version]
- Bossuyt, F.; Brown, R.M.; Hillis, D.M.; Cannatella, D.C.; Milinkovitch, M.C. Phylogeny and biogeography of a cosmopolitan frog radiation: Late Cretaceous diversification resulted in continent-scale endemism in the family Ranidae. Syst. Biol. 2006, 55, 579–594. [Google Scholar] [CrossRef]
- Voorhies, M.R.; Holman, J.A.; Xiang, X.X. The Hottell Ranch rhino quarries (basal Ogallala: Medial Barstovian), Banner County, Nebraska. Part I: Geological setting, faunal lists, lower vertebrates. Contrib. Geol. Univ. Wyo. 1987, 25, 55–69. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [Green Version]
- Pagel, M. Detecting correlated evolution on phylogenies: A general method for the comparative analysis of discrete characters. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1994, 255, 37–45. [Google Scholar]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, M.; Deng, L.; Xu, Z.; Shi, H.; Jia, X.; Lai, Z.; Ruan, Q.; Chen, W. Characteristics of the mitochondrial genome of Rana omeimontis and related species in Ranidae: Gene rearrangements and phylogenetic relationships. Ecol. Evol. 2020, 10, 12817–12837. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Pan, T.; Wu, G.; Kang, X.; Ali, I.; Zhou, W.; Li, J.; Wu, X.; Zhang, B. Species delimitation and evolutionary history of tree frogs in the Hyla chinensis group (Hylidae, Amphibian). Front. Ecol. Evol. 2020, 8, 234. [Google Scholar] [CrossRef]
- Okamura, S.; Arculus, R.J.; Martynov, Y.A. Cenozoic magmatism of the North-Eastern Eurasian margin: The role of lithosphere versus asthenosphere. J. Petrol. 2005, 46, 221–253. [Google Scholar] [CrossRef] [Green Version]
- Zachos, J.; Pagani, M.; Sloan, L.; Thomas, E.; Billups, K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Zachos, J.C.; Dickens, G.R.; Zeebe, R.E. An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics. Nature 2008, 451, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Irisarri, I.; San Mauro, D.; Abascal, F.; Ohler, A.; Vences, M.; Zardoya, R. The origin of modern frogs (Neobatrachia) was accompanied by acceleration in mitochondrial and nuclear substitution rates. Bmc Genom. 2012, 13, 626. [Google Scholar] [CrossRef] [Green Version]
- Igawa, T.; Kurabayashi, A.; Usuki, C.; Fujii, T.; Sumida, M. Complete mitochondrial genomes of three neobatrachian anurans: A case study of divergence time estimation using different data and calibration settings. Gene 2008, 407, 116–129. [Google Scholar] [CrossRef]
- Kurabayashi, A.; Sumida, M.; Yonekawa, H.; Glaw, F.; Vences, M.; Hasegawa, M. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol. Biol. Evol. 2008, 25, 874–891. [Google Scholar] [CrossRef] [Green Version]
- Dowton, M.; Castro, L.R.; Campbell, S.L.; Bargon, S.D.; Austin, A.D. Frequent mitochondrial gene rearrangements at the hymenopteran nad3–nad5 junction. J. Mol. Evol. 2003, 56, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Kurabayashi, A.; Yoshikawa, N.; Sato, N.; Hayashi, Y.; Oumi, S.; Fujii, T.; Sumida, M. Complete mitochondrial DNA sequence of the endangered frog Odorrana ishikawae (family Ranidae) and unexpected diversity of mt gene arrangements in ranids. Mol. Phylogenet. Evol. 2010, 56, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Sumida, M.; Kanamori, Y.; Kaneda, H.; Kato, Y.; Nishioka, M.; Hasegawa, M.; Yonekawa, H. Complete nucleotide sequence and gene rearrangement of the mitochondrial genome of the Japanese pond frog Rana nigromaculata. Genes Genet. Syst. 2001, 76, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Miao, G.; Hu, S.; Sun, Q.; Ding, H.; Ji, Z.; Guo, P.; Yan, S.; Wang, C.; Kan, X. Quantification and evolution of mitochondrial genome rearrangement in Amphibians. Bmc Ecol. Evol. 2021, 21, 19. [Google Scholar] [CrossRef]
- San Mauro, D.; Gower, D.J.; Zardoya, R.; Wilkinson, M. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol. Biol. Evol. 2006, 23, 227–234. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Abbreviations | Length(bp) | GenBank |
|---|---|---|---|
| Rana zhenhaiensis * | zhe | 18,006 | OL681880 |
| Rana longicrus * | lon | 17,502 | OL681879 |
| Rana huanrensis | hua | 19,253 | KT588071 |
| Rana amurensis | amu01 | 20,571 | MF370348 |
| Rana amurensis | amu02 | 18,470 | KU343216 |
| Rana kunyuensis | kun | 22,255 | KF840516 |
| Rana chensinensis | che | 18,808 | KF898356 |
| Rana dybowskii | dyb | 18,864 | KF898355 |
| Rana omeimontis | ome | 20,120 | MK483118 |
| Rana kukunoris | kuk | 18,863 | KU246049 |
| Rana chaochiaoensis | cha | 18,591 | KU246048 |
| Rana draytonii | dra | 17,805 | KP013110 |
| Rana catesbeiana | cat01 | 17,681 | KX686108 |
| Rana catesbeiana | cat02 | 17,682 | AB761267 |
| Rana catesbeiana | cat03 | 18,241 | KF049927 |
| Rana sylvatica | syl | 17,343 | KP222281 |
| Rana okaloosae | oka | 17,504 | KP013096 |
| Rana pyrenaica | pyr | 17,211 | KU720300 |
| Rana temporaria | tem | 16,061 | MH536744 |
| Rana uenoi | uen | 17,370 | MW009067 |
| Babina subaspera | Bsub | 18,525 | NC_022871 |
| Pelophylax nigromaculatus | Pnig | 17,567 | KT878718 |
| AA | Codon | Count | Percentage (%) | RSCU | AA | Codon | Count | Percentage (%) | RSCU |
|---|---|---|---|---|---|---|---|---|---|
| Phe (F) | UUU | 133.9 | 3.59 | 1.02 | Ala (A) | GCA | 69.9 | 1.87 | 0.87 |
| Phe (F) | UUC | 128.5 | 3.44 | 0.98 | Ala (A) | GCG | 13.4 | 0.36 | 0.17 |
| Leu2 (L2) | UUA | 119.8 | 3.21 | 1.15 | Tyr (Y) | UAU | 55.8 | 1.49 | 0.98 |
| Leu2 (L2) | UUG | 25.6 | 0.69 | 0.25 | Tyr (Y) | UAC | 58.4 | 1.56 | 1.02 |
| Leu1 (L1) | CUU | 135.2 | 3.62 | 1.3 | His (H) | CAU | 31.6 | 0.85 | 0.64 |
| Leu1 (L1) | CUC | 158.6 | 4.25 | 1.52 | His (H) | CAC | 66.9 | 1.79 | 1.36 |
| Leu1 (L1) | CUA | 143.8 | 3.85 | 1.38 | Gln (Q) | CAA | 75.5 | 2.02 | 1.78 |
| Leu1 (L1) | CUG | 43.4 | 1.16 | 0.42 | Gln (Q) | CAG | 9.3 | 0.25 | 0.22 |
| Ile (I) | AUU | 170.3 | 4.56 | 1.12 | Asn (N) | AAU | 60.1 | 1.61 | 0.94 |
| Ile (I) | AUC | 134.5 | 3.60 | 0.88 | Asn (N) | AAC | 67.6 | 1.81 | 1.06 |
| Met (M) | AUA | 115.2 | 3.09 | 1.42 | Lys (K) | AAA | 70.7 | 1.89 | 1.74 |
| Met (M) | AUG | 47 | 1.26 | 0.58 | Lys (K) | AAG | 10.8 | 0.29 | 0.26 |
| Val (V) | GUU | 57.4 | 1.54 | 1.17 | Asp (D) | GAU | 26.4 | 0.71 | 0.72 |
| Val (V) | GUC | 52.5 | 1.41 | 1.07 | Asp (D) | GAC | 46.6 | 1.25 | 1.28 |
| Val (V) | GUA | 61.5 | 1.65 | 1.25 | Glu (E) | GAA | 68.6 | 1.84 | 1.56 |
| Val (V) | GUG | 25.4 | 0.68 | 0.52 | Glu (E) | GAG | 19.5 | 0.52 | 0.44 |
| Ser2 (S2) | UCU | 63.2 | 1.69 | 1.43 | Cys (C) | UGU | 12.8 | 0.34 | 0.82 |
| Ser2 (S2) | UCC | 75.9 | 2.03 | 1.71 | Cys (C) | UGC | 18.3 | 0.49 | 1.18 |
| Ser2 (S2) | UCA | 66.6 | 1.78 | 1.5 | Trp (W) | UGA | 87.8 | 2.35 | 1.63 |
| Ser2 (S2) | UCG | 9.1 | 0.24 | 0.21 | Trp (W) | UGG | 19.8 | 0.53 | 0.37 |
| Pro (P) | CCU | 44.7 | 1.20 | 0.88 | Arg (R) | CGU | 12.1 | 0.32 | 0.65 |
| Pro (P) | CCC | 88.2 | 2.36 | 1.73 | Arg (R) | CGC | 22.6 | 0.61 | 1.23 |
| Pro (P) | CCA | 59.6 | 1.60 | 1.17 | Arg (R) | CGA | 32.8 | 0.88 | 1.78 |
| Pro (P) | CCG | 11.1 | 0.30 | 0.22 | Arg (R) | CGG | 6.2 | 0.17 | 0.34 |
| Thr (T) | ACU | 72.4 | 1.94 | 1.01 | Ser (S1) | AGU | 16.4 | 0.44 | 0.37 |
| Thr (T) | ACC | 102.2 | 2.74 | 1.43 | Ser (S1) | AGC | 34.6 | 0.93 | 0.78 |
| Thr (T) | ACA | 98.8 | 2.65 | 1.38 | Gly (G) | GGU | 28.8 | 0.77 | 0.52 |
| Thr (T) | ACG | 13.1 | 0.35 | 0.18 | Gly (G) | GGC | 81.5 | 2.18 | 1.47 |
| Ala (A) | GCU | 86.8 | 2.33 | 1.08 | Gly (G) | GGA | 64.7 | 1.73 | 1.17 |
| Ala (A) | GCC | 152.6 | 4.09 | 1.89 | Gly (G) | GGG | 46.9 | 1.26 | 0.85 |
| Gene | Length (bp) | %Vs | %Pis | %S | ts/tv | Ks | Ka | Ka/Ks | AT% | AT- Skew | GC- Skew | Aupd |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ND1 | 960 | 48.02 | 41.67 | 6.35 | 4.26 | 0.98 | 0.08 | 0.08 | 56.24 | −0.14 | −0.41 | 0.12 |
| ND2 | 1032 | 48.45 | 40.79 | 7.66 | 4.19 | 0.85 | 0.08 | 0.09 | 57.2 | −0.03 | −0.47 | 0.13 |
| COI | 1551 | 36.56 | 32.17 | 4.38 | 5.16 | 0.93 | 0.02 | 0.02 | 54.83 | −0.09 | −0.21 | 0.02 |
| COII | 687 | 35.66 | 30.71 | 4.95 | 5.99 | 0.78 | 0.03 | 0.04 | 56.53 | 0.04 | −0.28 | 0.05 |
| ATP8 | 159 | 54.09 | 42.14 | 11.95 | 5.79 | 0.55 | 0.1 | 0.18 | 61.89 | 0.04 | −0.47 | 0.21 |
| ATP6 | 693 | 45.74 | 38.67 | 7.07 | 5.27 | 0.96 | 0.06 | 0.06 | 57.35 | −0.11 | −0.47 | 0.09 |
| COIII | 783 | 35.50 | 30.27 | 5.24 | 5.35 | 0.8 | 0.03 | 0.04 | 53.58 | −0.14 | −0.28 | 0.05 |
| ND3 | 339 | 50.44 | 44.84 | 5.60 | 4.26 | 0.53 | 0.09 | 0.17 | 54.87 | −0.22 | −0.36 | 0.15 |
| ND4L | 282 | 50.00 | 46.43 | 8.51 | 4.50 | 0.53 | 0.08 | 0.15 | 55.84 | −0.11 | −0.40 | 0.12 |
| ND4 | 1359 | 44.81 | 37.97 | 6.84 | 4.31 | 0.92 | 0.06 | 0.07 | 56.81 | −0.08 | −0.41 | 0.09 |
| ND5 | 1740 | 57.99 | 47.41 | 10.57 | 3.46 | 1.08 | 0.14 | 0.13 | 56.71 | −0.09 | −0.34 | 0.20 |
| Cyt b | 1140 | 39.56 | 33.51 | 6.05 | 4.18 | 1.00 | 0.03 | 0.03 | 54.00 | −0.10 | −0.4 | 0.05 |
| ND6 | 486 | 50.41 | 43.62 | 6.80 | 3.98 | 0.84 | 0.12 | 0.14 | 52.81 | −0.31 | 0.54 | 0.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Qian, W.; Miao, K.; Qian, R.; Yuan, S.; Liu, W.; Dai, J.; Hu, C.; Chang, Q. Comparative Mitogenomics of True Frogs (Ranidae, Anura), and Its Implications for the Phylogeny and Evolutionary History of Rana. Animals 2022, 12, 1250. https://doi.org/10.3390/ani12101250
Chen W, Qian W, Miao K, Qian R, Yuan S, Liu W, Dai J, Hu C, Chang Q. Comparative Mitogenomics of True Frogs (Ranidae, Anura), and Its Implications for the Phylogeny and Evolutionary History of Rana. Animals. 2022; 12(10):1250. https://doi.org/10.3390/ani12101250
Chicago/Turabian StyleChen, Wan, Weiya Qian, Keer Miao, Ruen Qian, Sijia Yuan, Wei Liu, Jianhua Dai, Chaochao Hu, and Qing Chang. 2022. "Comparative Mitogenomics of True Frogs (Ranidae, Anura), and Its Implications for the Phylogeny and Evolutionary History of Rana" Animals 12, no. 10: 1250. https://doi.org/10.3390/ani12101250
APA StyleChen, W., Qian, W., Miao, K., Qian, R., Yuan, S., Liu, W., Dai, J., Hu, C., & Chang, Q. (2022). Comparative Mitogenomics of True Frogs (Ranidae, Anura), and Its Implications for the Phylogeny and Evolutionary History of Rana. Animals, 12(10), 1250. https://doi.org/10.3390/ani12101250