
Identification of Genomic Regions and Candidate Genes for Litter Traits in French Large White Pigs Using Genome-Wide Association Studies

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Animal and Phenotypic Data
2.2. Genomic Data and Quality Control
2.3. Estimation of Genetic Parameters
2.4. Genome-Wide Association Study
2.5. Single-Step Genome-Wide Association Study
2.6. Annotation of Genomic Regions and Search for Genes
3. Results
3.1. Phenotypic Statistics
3.2. Genetic Parameter Statistics
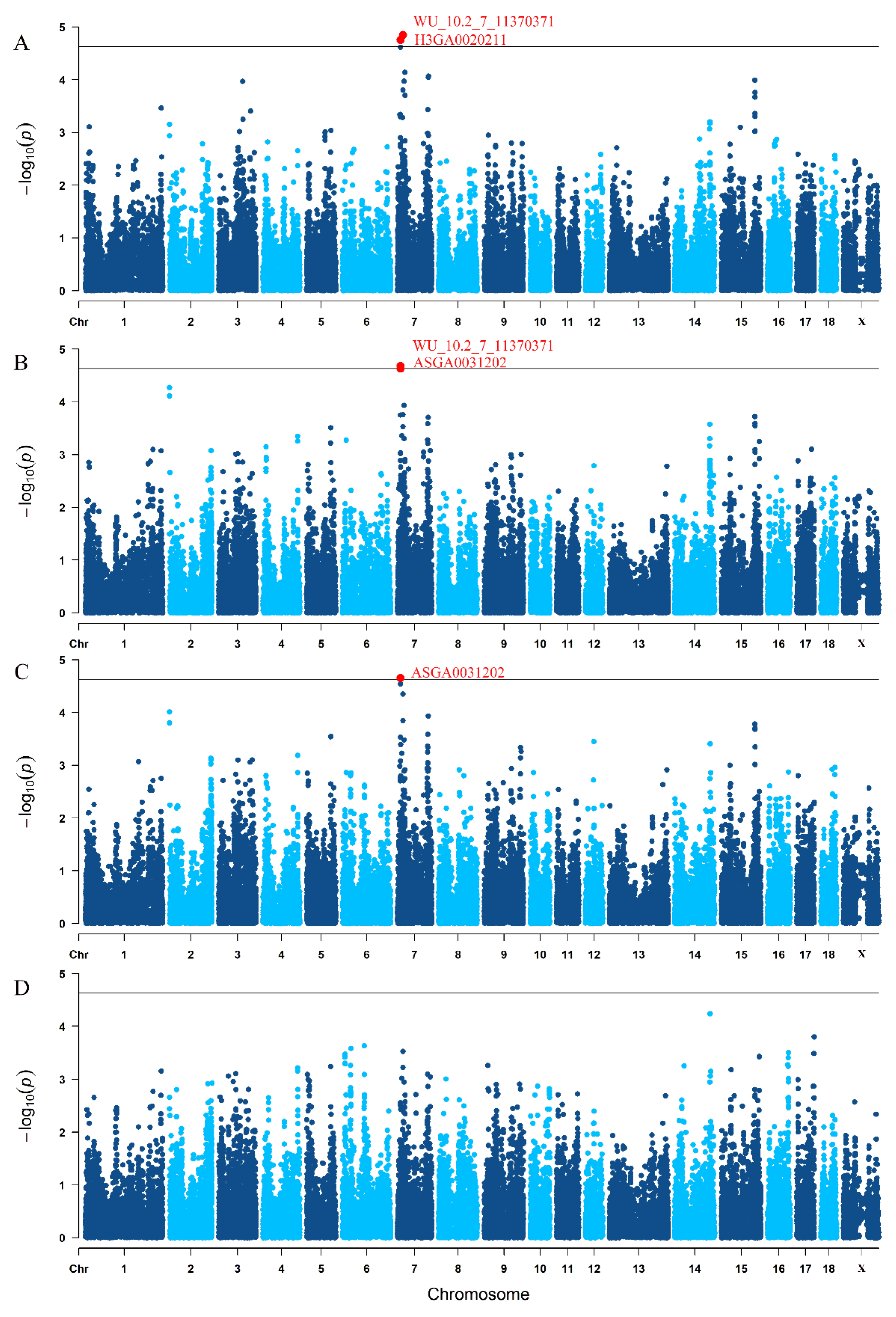
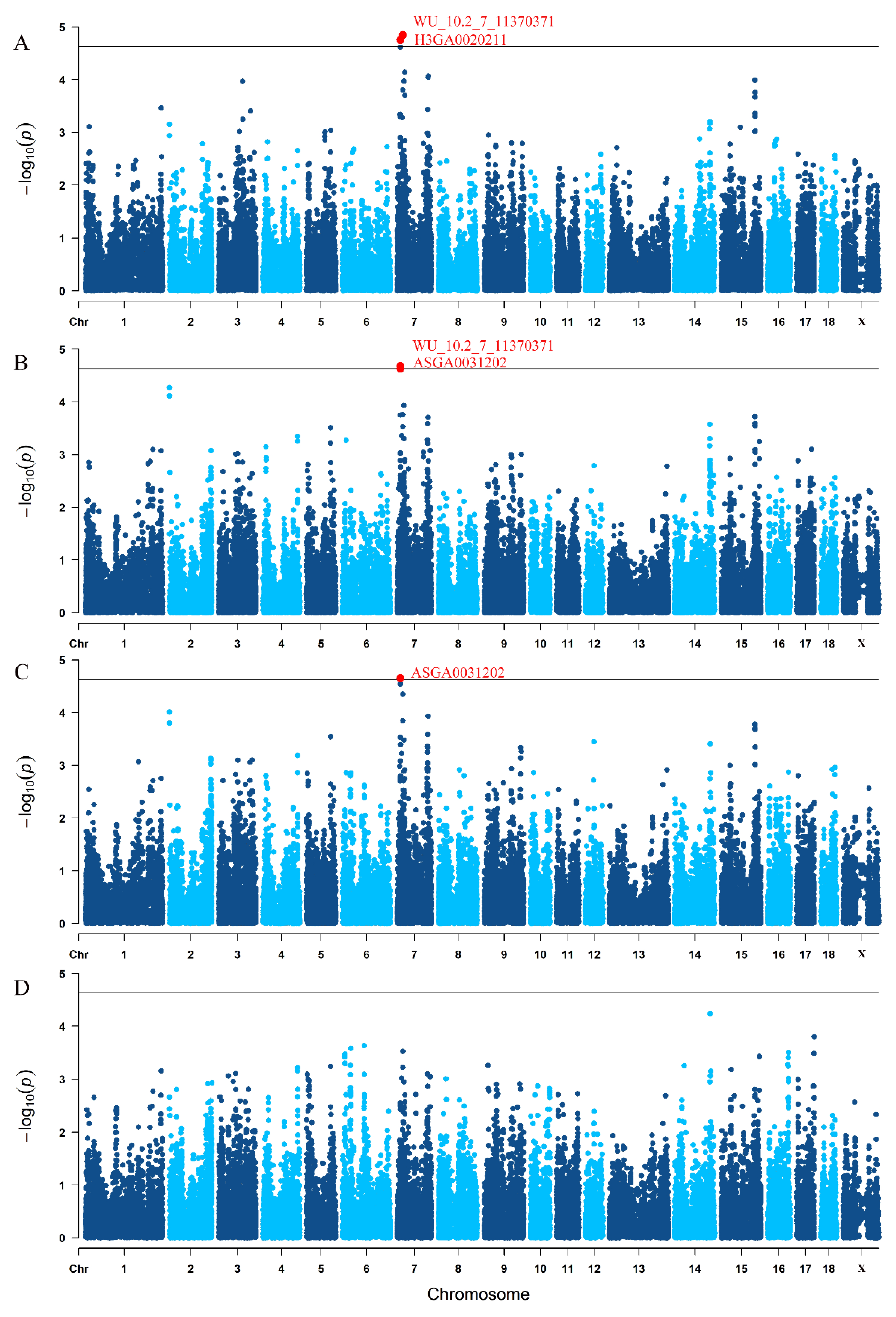
3.3. Traditional GWAS for Litter Traits
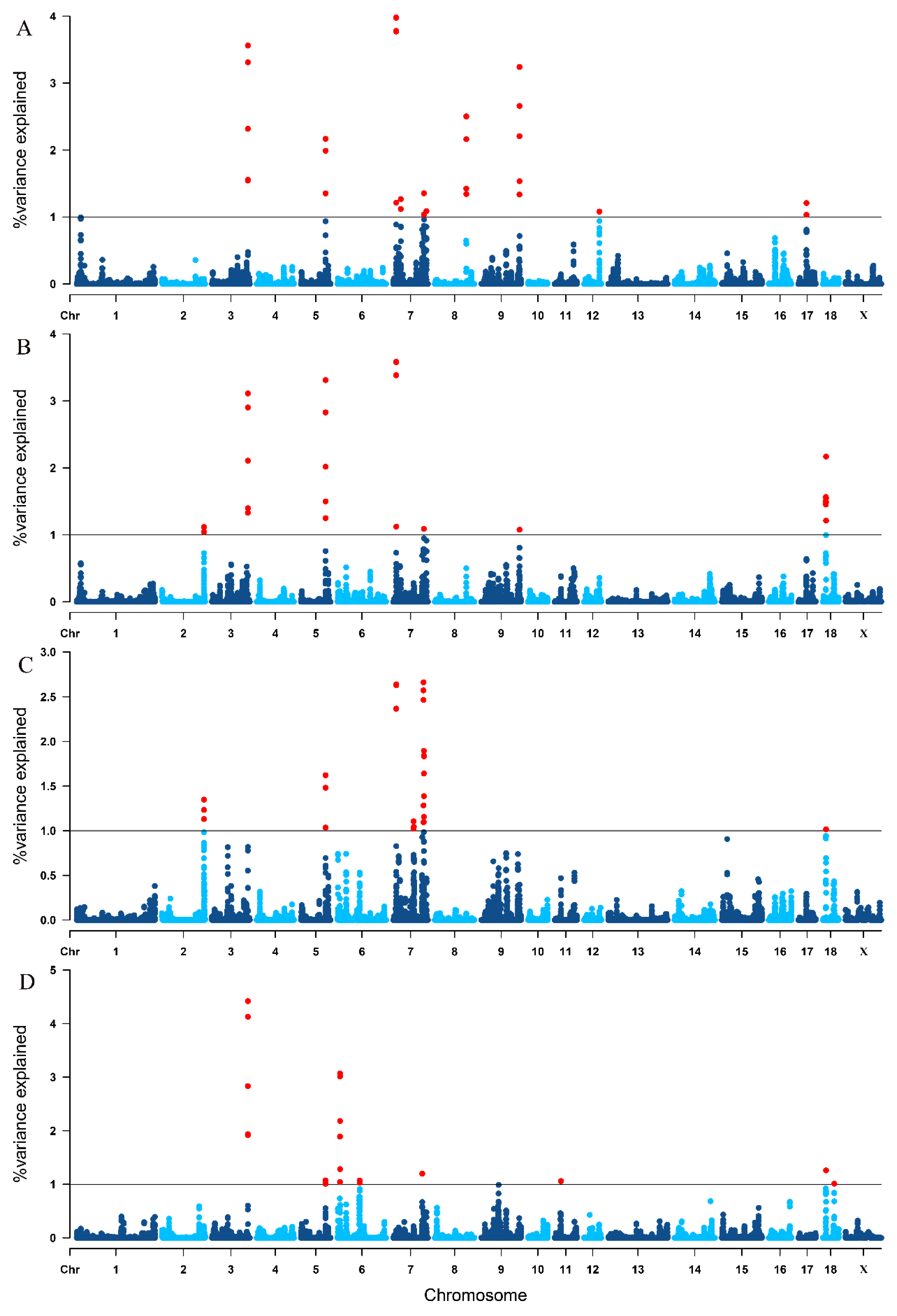
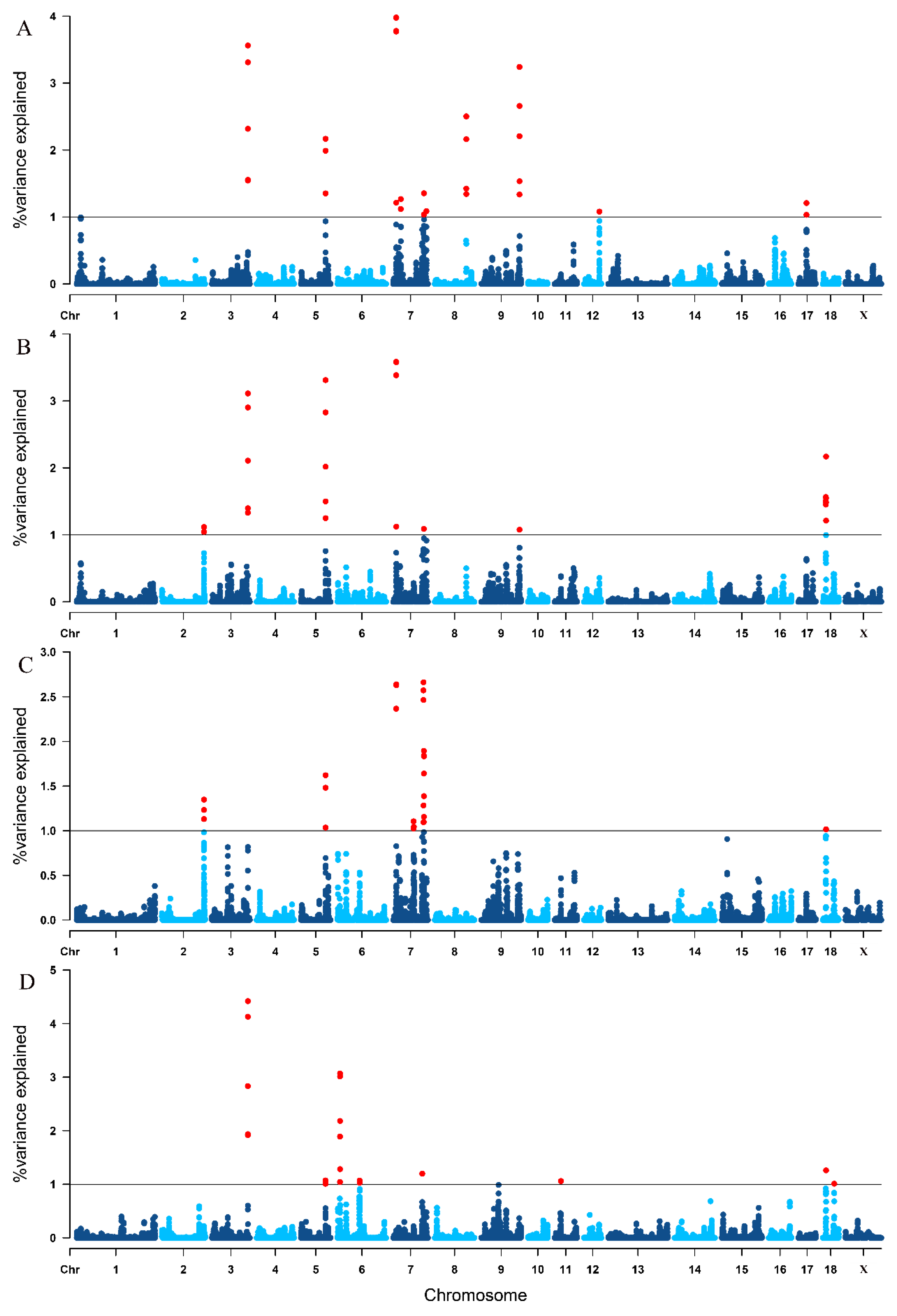
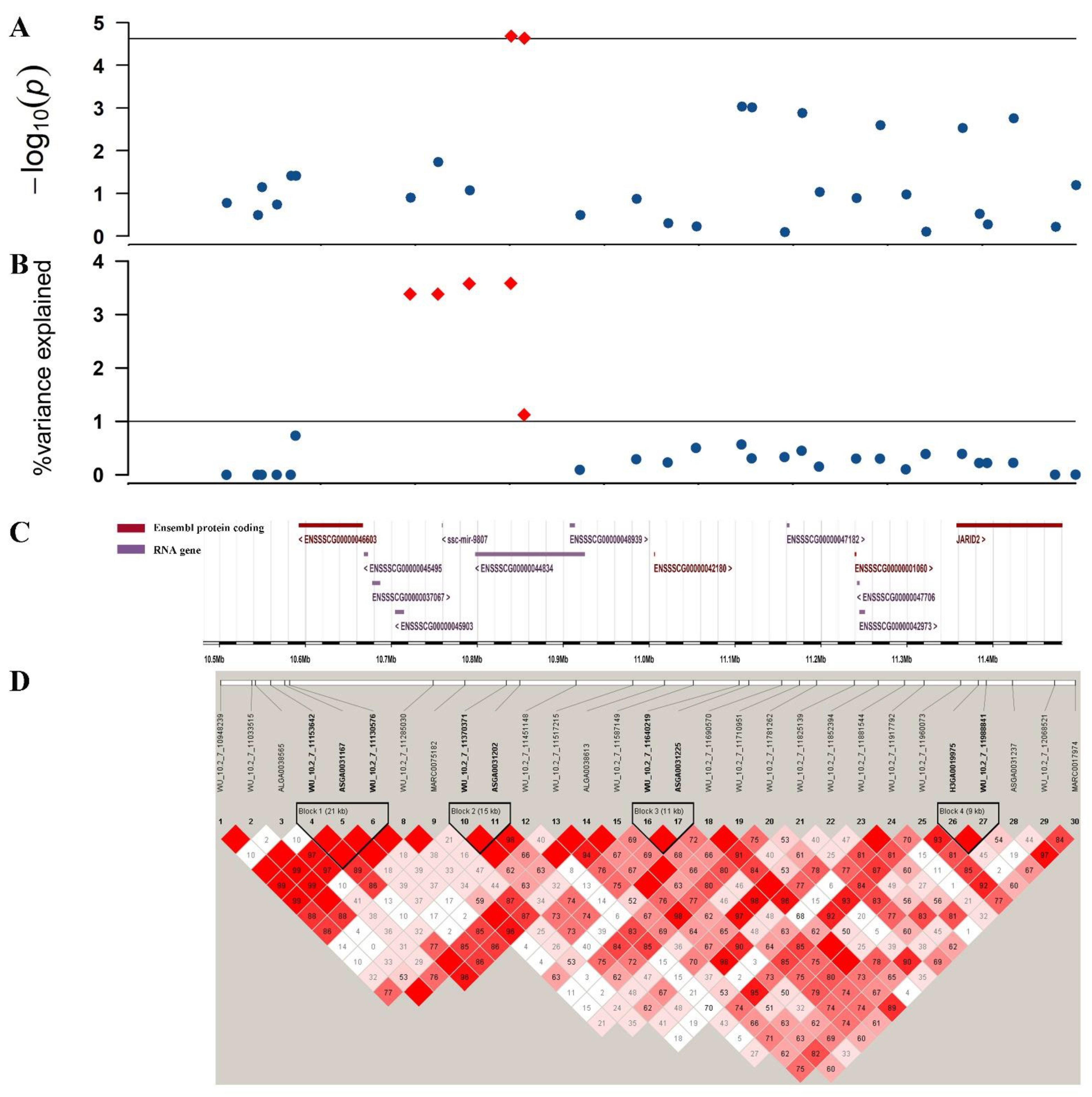
3.4. Single-Step GWAS for Litter Traits
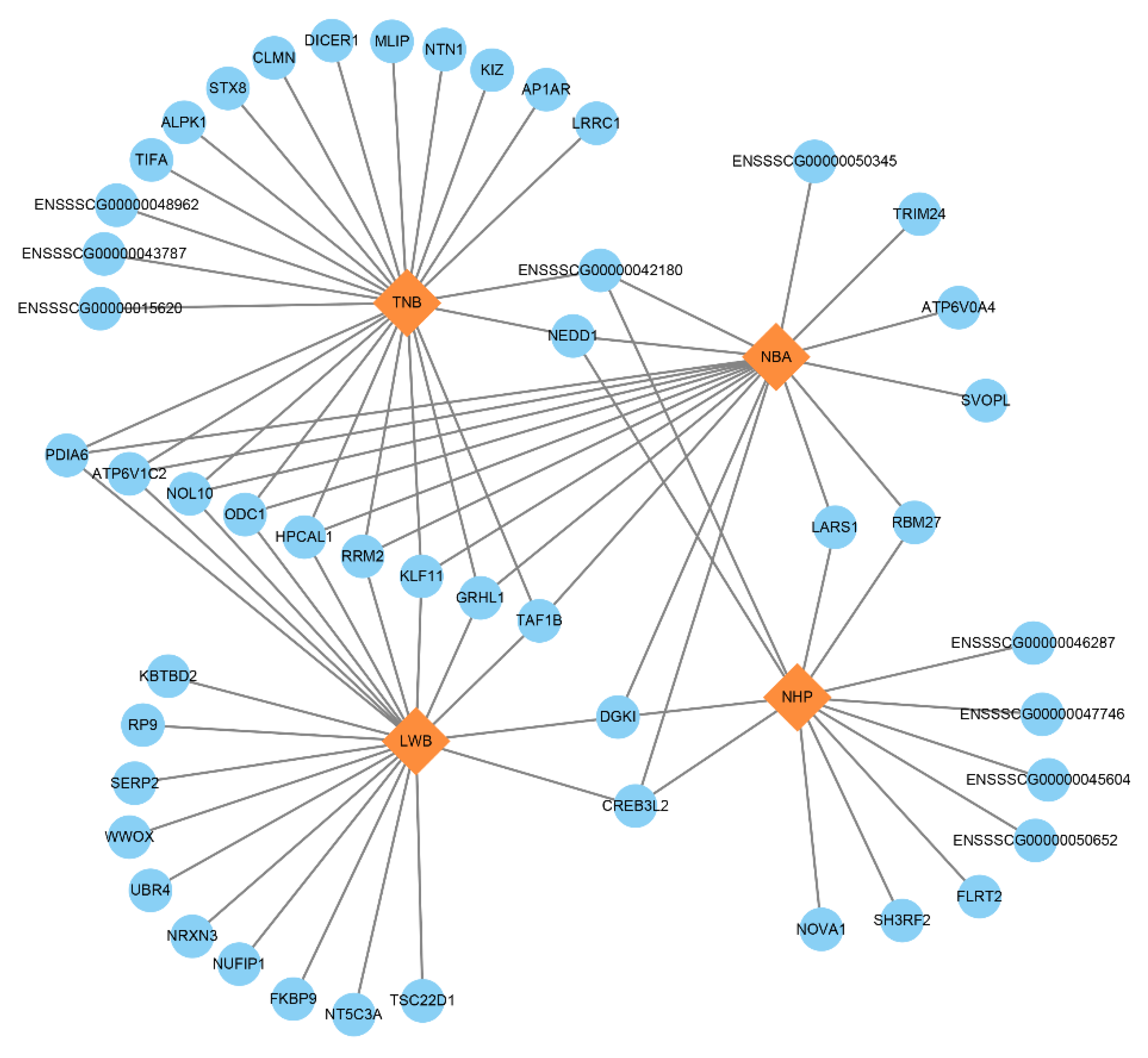
3.5. Identification of Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ye, J.; Tan, C.; Hu, X.; Wang, A.; Wu, Z. Genetic parameters for reproductive traits at different parities in Large White pigs. J. Anim. Sci. 2018, 96, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Konta, A.; Kimata, M.; Ishii, K.; Uemoto, Y.; Satoh, M. Estimation of genetic parameters for farrowing traits in purebred Landrace and Large White pigs. Anim. Sci. J. 2019, 90, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canario, L.; Roy, N.; Gruand, J.; Bidanel, J.P. Genetic variation of farrowing kinetics traits and their relationships with litter size and perinatal mortality in French Large White sows. J. Anim. Sci. 2006, 84, 1053–1058. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Zhang, J.; Jiang, Y.; Gao, H.; Tang, S.; Mi, S.; Yu, F.; Meng, Q.; Xiao, W.; Zhang, Q.; et al. Genomic prediction for growth and reproduction traits in pig using an admixed reference population. J. Anim. Sci. 2017, 95, 3415–3424. [Google Scholar] [CrossRef]
- Ding, R.; Qiu, Y.; Zhuang, Z.; Ruan, D.; Wu, J.; Zhou, S.; Ye, J.; Cao, L.; Hong, L.; Xu, Z.; et al. Genome-wide association studies reveals polygenic genetic architecture of litter traits in Duroc pigs. Theriogenology 2021, 173, 269–278. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Zhong, Z.; Jiang, S. A Whole Genome Sequencing-Based Genome-Wide Association Study Reveals the Potential Associations of Teat Number in Qingping Pigs. Animals 2022, 12, 1057. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Duan, D.; Xue, Y.; Han, X.; Wang, K.; Qiao, R.; Li, X.-L.; Li, X.-J. An association study on imputed whole-genome resequencing from high-throughput sequencing data for body traits in crossbred pigs. Anim. Genet. 2022, 53, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Li, C.; Duan, D.; Wang, M.; Han, X.; Wang, K.; Qiao, R.; Li, X.J.; Li, X.L. Genome-wide association studies for growth-related traits in a crossbreed pig population. Anim. Genet. 2021, 52, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, M.; Jacobson, C.; Vaske, D.; Tuggle, C.; Wang, L.; Short, T.; Eckardt, G.; Sasaki, S.; Vincent, A.; McLaren, D.; et al. The estrogen receptor locus is associated with a major gene influencing litter size in pigs. Proc. Natl. Acad. Sci. USA 1996, 93, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.L.; Wang, L.; Tuggle, C.K.; Robic, A.; Rothschild, M.F. Prolactin receptor maps to pig chromosome 16. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 1997, 8, 793–794. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, N.; Xiao, L.; Cao, G.; Chen, Y.; Zhang, S.; Chen, Y.; Wu, C.; Zhang, J.; Sun, S.; et al. FSHB subunit gene is associated with major gene controlling litter size in commercial pig breeds. Sci. China Ser. C Life Sci. 1998, 41, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.X.; Huang, Y.S.; Ye, J.; Zhang, W.; Li, Y.; Nie, Q.H. Identification and characterization of RFRP gene in pigs and its association with reproductive traits. Genet. Mol. Res. GMR 2014, 13, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Yang, Q.; Wang, K.; Zhou, J.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W.; Jiang, A.; Jiang, Y.; et al. Single step genome-wide association studies based on genotyping by sequence data reveals novel loci for the litter traits of domestic pigs. Genomics 2018, 110, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Xu, Y.; Ju, J.Q.; Pan, Z.N.; Liu, J.C.; Sun, S.C. Increased Environment-Related Metabolism and Genetic Expression in the In Vitro Matured Mouse Oocytes by Transcriptome Analysis. Front. Cell Dev. Biol. 2021, 9, 642010. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet Res 2012, 94, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Sell-Kubiak, E.; Knol, E.F.; Lopes, M. Evaluation of the phenotypic and genomic background of variability based on litter size of Large White pigs. Genet. Sel. Evol. 2022, 54, 1. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Misztal, I.; Tsuruta, S.; Strabel, T.; Auvray, B.; Druet, T.; Lee, D.H. BLUPF90 and related programs (BGF90). In Proceedings of the 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France, 19–23 August 2002. [Google Scholar]
- Legarra, A.; Aguilar, I.; Misztal, I. A relationship matrix including full pedigree and genomic information. J. Dairy Sci. 2009, 92, 4656–4663. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Liu, X. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated tool for Genome-Wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–629. [Google Scholar] [CrossRef]
- Beissinger, T.M.; Rosa, G.J.; Kaeppler, S.M.; Gianola, D.; de Leon, N. Defining window-boundaries for genomic analyses using smoothing spline techniques. Genet. Sel. Evol. 2015, 47, 30. [Google Scholar] [CrossRef] [Green Version]
- Suwannasing, R.; Duangjinda, M.; Boonkum, W.; Taharnklaew, R.; Tuangsithtanon, K. The identification of novel regions for reproduction trait in Landrace and Large White pigs using a single step genome-wide association study. Asian-Australas. J. Anim. Sci. 2018, 31, 1852–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2019, 47, D701–D710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Xu, J.J.; Sun, X.L.; Jiang, H.; Han, D.X.; Liu, C.; Gao, Y.; Yuan, B.; Zhang, J.B. Function of JARID2 in bovines during early embryonic development. PeerJ 2017, 5, e4189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landeira, D.; Sauer, S.; Poot, R.; Dvorkina, M.; Mazzarella, L.; Jorgensen, H.F.; Pereira, C.F.; Leleu, M.; Piccolo, F.M.; Spivakov, M.; et al. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat. Cell Biol. 2010, 12, 618–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, D.; Cloos, P.A.; Walfridsson, J.; Olsson, L.; Bukowski, J.P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010, 464, 306–310. [Google Scholar] [CrossRef]
- Ma, X.S.; Lin, F.; Wang, Z.W.; Hu, M.W.; Huang, L.; Meng, T.G.; Jiang, Z.Z.; Schatten, H.; Wang, Z.B.; Sun, Q.Y. Geminin deletion in mouse oocytes results in impaired embryo development and reduced fertility. Mol. Biol. Cell 2016, 27, 768–775. [Google Scholar] [CrossRef]
- Yuan, Y.; Wheeler, M.B.; Krisher, R.L. Disrupted redox homeostasis and aberrant redox gene expression in porcine oocytes contribute to decreased developmental competence. Biol. Reprod. 2012, 87, 78. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.R.D.; Jain, S.; Banerjee, A. Expression of ODC1, SPD, SPM and AZIN1 in the hypothalamus, ovary and uterus during rat estrous cycle. Gen. Comp. Endocrinol. 2017, 246, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Tabbaa, Z.M.; Khan, Z.; Schoolmeester, J.K.; El-Nashar, S.; Famuyide, A.; Keeney, G.L.; Daftary, G.S. Epigenetic regulation of uterine biology by transcription factor KLF11 via posttranslational histone deacetylation of cytochrome p450 metabolic enzymes. Endocrinology 2014, 155, 4507–4520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taracha, A.; Kotarba, G.; Wilanowski, T. Neglected Functions of TFCP2/TFCP2L1/UBP1 Transcription Factors May Offer Valuable Insights into Their Mechanisms of Action. Int. J. Mol. Sci. 2018, 19, 2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Brito, L.F.; Luo, H.; Shi, R.; Chang, Y.; Liu, L.; Guo, G.; Wang, Y. Genetic and Genomic Analyses of Service Sire Effect on Female Reproductive Traits in Holstein Cattle. Front. Genet. 2021, 12, 713575. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, T.; Kim, S.T.; Zakrzewska, A.; Lee, B.E.; Kang, M.J.; Yoo, Y.D.; Cha-Molstad, H.J.; Hwang, J.; Soung, N.K.; Sung, K.S.; et al. UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 3800–3805. [Google Scholar] [CrossRef] [Green Version]
- Bergfelder-Druing, S.; Grosse-Brinkhaus, C.; Lind, B.; Erbe, M.; Schellander, K.; Simianer, H.; Tholen, E. A genome-wide association study in large white and landrace pig populations for number piglets born alive. PLoS ONE 2015, 10, e0117468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, M.M.; Najmula, J.; Guzewska, M.M.; Przygrodzka, E. MiRNAs in the Peri-Implantation Period: Contribution to Embryo-Maternal Communication in Pigs. Int. J. Mol. Sci. 2020, 21, 2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrola-Barraza, M.E.; Hernandez-Seanez, R.; Barcelo-Fimbres, M.; Rodriguez-Almeida, F.A.; Gonzalez-Rodriguez, E.; Garcia-Quinonez, S.; Grado-Ahuir, J.A.; Moreno-Brito, V. Dicer gene expression during early bovine embryo development. Mol. Reprod. Dev. 2011, 78, 622. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer is essential for mouse development. Nat. Genet. 2003, 35, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Marzinke, M.A.; Henderson, E.M.; Yang, K.S.; See, A.W.; Knutson, D.C.; Clagett-Dame, M. Calmin expression in embryos and the adult brain, and its regulation by all-trans retinoic acid. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2010, 239, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Rosendo, A.; Druet, T.; Gogue, J.; Canario, L.; Bidanel, J.P. Correlated responses for litter traits to six generations of selection for ovulation rate or prenatal survival in French Large White pigs. J. Anim. Sci. 2007, 85, 1615–1624. [Google Scholar] [CrossRef]
- Wolf, J.; Žáková, E.; Groeneveld, E. Within-litter variation of birth weight in hyperprolific Czech Large White sows and its relation to litter size traits, stillborn piglets and losses until weaning. Livest. Sci. 2008, 115, 195–205. [Google Scholar] [CrossRef]
- Li, K.; Ren, J.; Xing, Y.; Zhang, Z.; Ma, J.; Guo, Y.; Huang, L. Quantitative trait loci for litter size and prenatal loss in a White Duroc x Chinese Erhualian resource population. Anim. Genet. 2009, 40, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, P.H.; Zhu, M.X.; He, L.C.; Sui, S.P.; Gao, S.; Su, G.S.; Ding, N.S.; Huang, Y.; Lu, Z.Q.; et al. Genome-wide association analysis reveals genomic regions on Chromosome 13 affecting litter size and candidate genes for uterine horn length in Erhualian pigs. Anim. Int. J. Anim. Biosci. 2018, 12, 2453–2461. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, S.C.; Finlayson, H.A.; Ashworth, C.J.; Haley, C.S.; Archibald, A.L. A genome-wide linkage analysis for reproductive traits in F2 Large White x Meishan cross gilts. Anim. Genet. 2014, 45, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.S.; Schulz, R.; Maretto, S.; Costello, I.; Srinivas, S.; Bikoff, E.; Robertson, E. The fibronectin leucine-rich repeat transmembrane protein Flrt2 is required in the epicardium to promote heart morphogenesis. Development 2011, 138, 1297–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait 1 | Mean | SD | Min | Max | CV 2 (%) |
|---|---|---|---|---|---|
| TNB | 14.22 | 3.88 | 3 | 25 | 27.32% |
| NBA | 12.77 | 3.57 | 3 | 23 | 27.97% |
| NHP | 12.00 | 3.37 | 1 | 23 | 28.10% |
| LWB (kg) | 15.31 | 4.21 | 2.6 | 27.9 | 27.51% |
| Trait 1 | TNB | NBA | NHP | LWB (kg) |
|---|---|---|---|---|
| TNB | 0.09 (0.04) | 0.82 (0.83) | 0.78 (1.87) | 0.38 (0.49) |
| NBA | 0.85 (0.01) | 0.06 (0.04) | 0.99 (0.31) | 0.70 (0.62) |
| NHP | 0.78 (0.01) | 0.93 (0.01) | 0.02 (0.03) | 0.94 (0.69) |
| LWB (kg) | 0.68 (0.01) | 0.83 (0.01) | 0.85 (0.01) | 0.13 (0.05) |
| Trait 1 | SNP | SSC 2 | Position (Mbp) | p-Value |
|---|---|---|---|---|
| TNB | WU_10.2_7_11370371 | 7 | 10.841911 | 1.77 × 10−5 |
| H3GA0020211 | 7 | 19.658767 | 1.42 × 10−5 | |
| NBA | WU_10.2_7_11370371 | 7 | 10.841911 | 2.09 × 10−5 |
| ASGA0031202 | 7 | 10.857509 | 2.33 × 10−5 | |
| NHP | ASGA0031202 | 7 | 10.857509 | 2.21 × 10−5 |
| Trait 1 | SSC 2 | Genomic Region (Mbp) | Var (%) 3 |
|---|---|---|---|
| TNB | 3 | 125.756195–126.562853 | 3.561 |
| 5 | 86.486236–86.726806 | 2.168 | |
| 7 | 10.724105–11.058567 | 3.979 | |
| 7 | 7:26745500–27045611 | 1.268 | |
| 7 | 107.611784–107.768080 | 1.354 | |
| 7 | 116.391085–116.501529 | 1.087 | |
| 8 | 110.219894–110.662629 | 2.503 | |
| 9 | 133.809598–134.161791 | 3.241 | |
| 12 | 54.264915–54.434349 | 1.080 | |
| 17 | 28.878576–29.033480 | 1.208 | |
| NBA | 2 | 147.530240–147.685182 | 1.117 |
| 3 | 125.756195–126.562853 | 3.112 | |
| 5 | 86.486236–86.784197 | 3.311 | |
| 7 | 10.724105–11.058567 | 3.585 | |
| 7 | 107.611784–107.730536 | 1.089 | |
| 9 | 133.948708–134.086750 | 1.076 | |
| 18 | 10.909022–11.384836 | 1.565 | |
| 18 | 11.574718–11.910292 | 2.170 | |
| NHP | 2 | 147.400783–147.421494 | 1.131 |
| 2 | 147.530240–147.685182 | 1.348 | |
| 5 | 86.486236–86.726806 | 1.621 | |
| 7 | 10.724105–11.025466 | 2.640 | |
| 7 | 71.250276–71.941528 | 1.105 | |
| 7 | 72.437167–72.997611 | 1.044 | |
| 7 | 105.990032–106.291677 | 2.663 | |
| 7 | 107.256824–107.401244 | 1.097 | |
| 7 | 107.589360–107.819775 | 1.895 | |
| 18 | 11.653824–11.778406 | 1.016 | |
| LWB | 3 | 125.756195–126.562853 | 4.420 |
| 5 | 86.178117–86.367402 | 1.072 | |
| 6 | 8.566888–9.033493 | 3.069 | |
| 6 | 77.495951–77.642338 | 1.071 | |
| 7 | 101.840965–102.117118 | 1.198 | |
| 11 | 22.269146–22.714741 | 1.060 | |
| 18 | 11.653824–11.778406 | 1.260 | |
| 18 | 40.459198–40.635413 | 1.011 |
| Method | Symbol | SSC 1 | Position (Mbp) | Trait | Species | Publication |
|---|---|---|---|---|---|---|
| GWAS | JARID2 | 7 | 11.357961–11.602104 | Early embryonic development | Bovines/mouse/pig | (Fu et al., 2017 [25]; Landeira et al., 2010 [26]; pasini et al., 2010 [27]) |
| GMNN | 7 | 19.585999–19.599879 | Embryo development and implantation | Mouse | (Ma et al., 2016 [28]) | |
| ssGWAS | PDIA6 | 3 | 125.751970–125.776955 | Oocyte quality | pig | (Yuan et al., 2012 [29]) |
| ODC1 | 3 | 126.078418–126.132577 | Oocyte physiology | Rat | (Fernandes et al., 2017 [30]) | |
| KLF11 | 3 | 126.418996–126.429904 | Uterine biology | Human | (Zheng et al., 2014 [31]) | |
| GRHL1 | 3 | 126.464198–126.511211 | Placenta development | Human | (Taracha et al., 2018 [32]) | |
| WWOX | 6 | 8.911139–9.871102 | Calving | dairy cattle | (Chen et al., 2021 [33]) | |
| UBR4 | 6 | 77.568415–77.707983 | Yolk sac vascular development | Mice | (Tasaki et al., 2013 [34]) | |
| FLRT2 | 7 | 107.812613–107.913618 | Embryo development | Pig | (Bergfelder et al., 2015 [35]) | |
| DICER1 | 7 | 116.361630–116.436519 | Embryo development | Pig/cattle/mouse | (Kaczmarek et al., 2020 [36]; Burrola-Barraza et al., 2011 [37]; Bernstein et al., 2003 [38]) | |
| CLMN | 7 | 116.476791–116.590265 | Embryo development | Human/mouse | (Marzinke et al., 2010 [39]) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Wu, Z.; Chen, R.; Huang, Z.; Han, X.; Qiao, R.; Wang, K.; Yang, F.; Li, X.-J.; Li, X.-L. Identification of Genomic Regions and Candidate Genes for Litter Traits in French Large White Pigs Using Genome-Wide Association Studies. Animals 2022, 12, 1584. https://doi.org/10.3390/ani12121584
Chen J, Wu Z, Chen R, Huang Z, Han X, Qiao R, Wang K, Yang F, Li X-J, Li X-L. Identification of Genomic Regions and Candidate Genes for Litter Traits in French Large White Pigs Using Genome-Wide Association Studies. Animals. 2022; 12(12):1584. https://doi.org/10.3390/ani12121584
Chicago/Turabian StyleChen, Jianmei, Ziyi Wu, Ruxue Chen, Zhihui Huang, Xuelei Han, Ruimin Qiao, Kejun Wang, Feng Yang, Xin-Jian Li, and Xiu-Ling Li. 2022. "Identification of Genomic Regions and Candidate Genes for Litter Traits in French Large White Pigs Using Genome-Wide Association Studies" Animals 12, no. 12: 1584. https://doi.org/10.3390/ani12121584
APA StyleChen, J., Wu, Z., Chen, R., Huang, Z., Han, X., Qiao, R., Wang, K., Yang, F., Li, X.-J., & Li, X.-L. (2022). Identification of Genomic Regions and Candidate Genes for Litter Traits in French Large White Pigs Using Genome-Wide Association Studies. Animals, 12(12), 1584. https://doi.org/10.3390/ani12121584