
Phylogenetic Implication of Large Intergenic Spacers: Insights from a Mitogenomic Comparison of Prosopocoilus Stag Beetles (Coleoptera: Lucanidae)


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Polymerase Chain Reaction (PCR) Amplification and Sequencing
2.3. Mitogenome Assembly, Annotation, and Analysis
2.4. Phylogenetic Analyses
3. Results and Discussion
3.1. Genome Organization and Base Composition
3.2. Protein-Coding Genes and Codon Usage
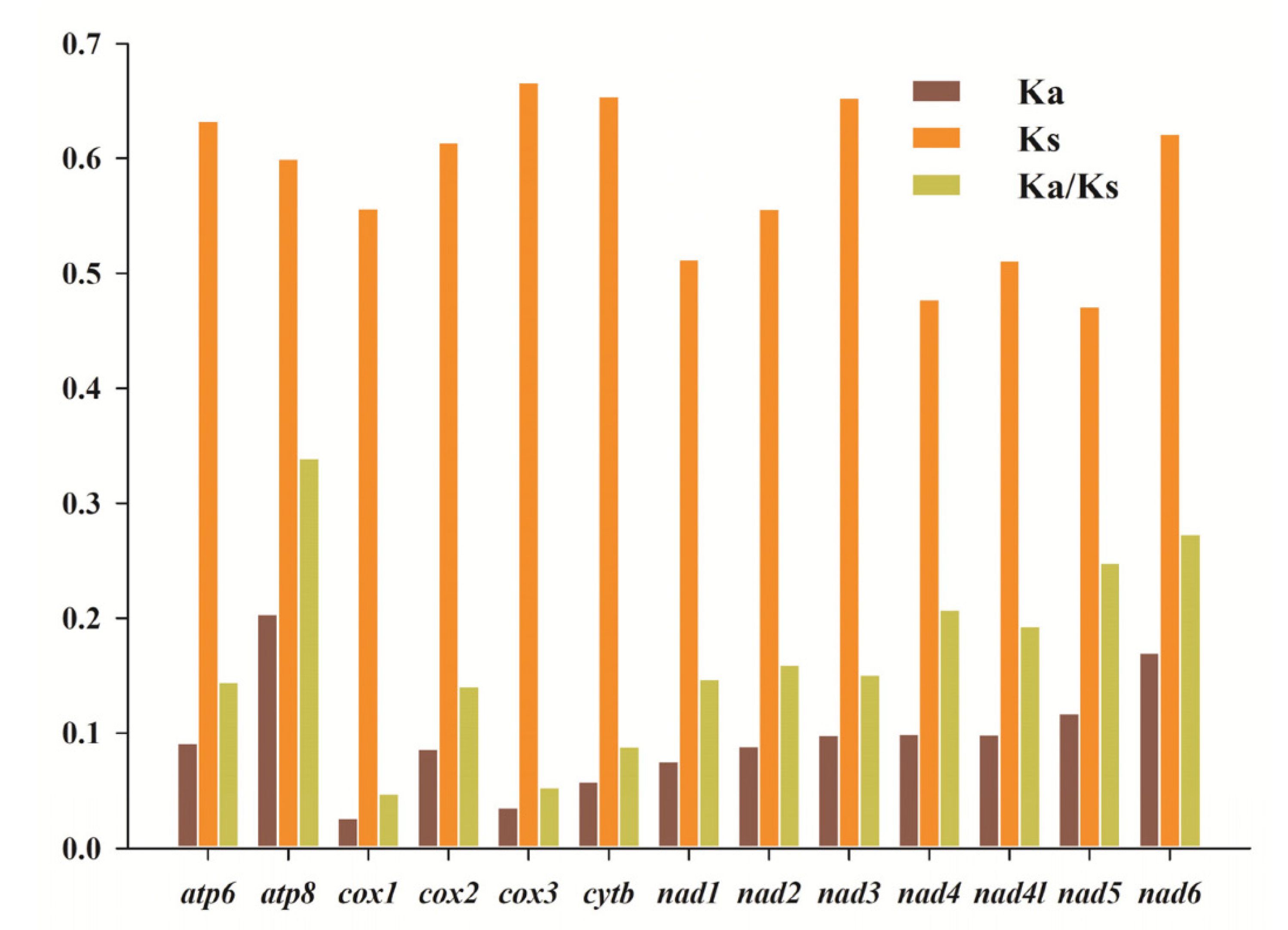
3.3. Evolutionary Rates of PCGs
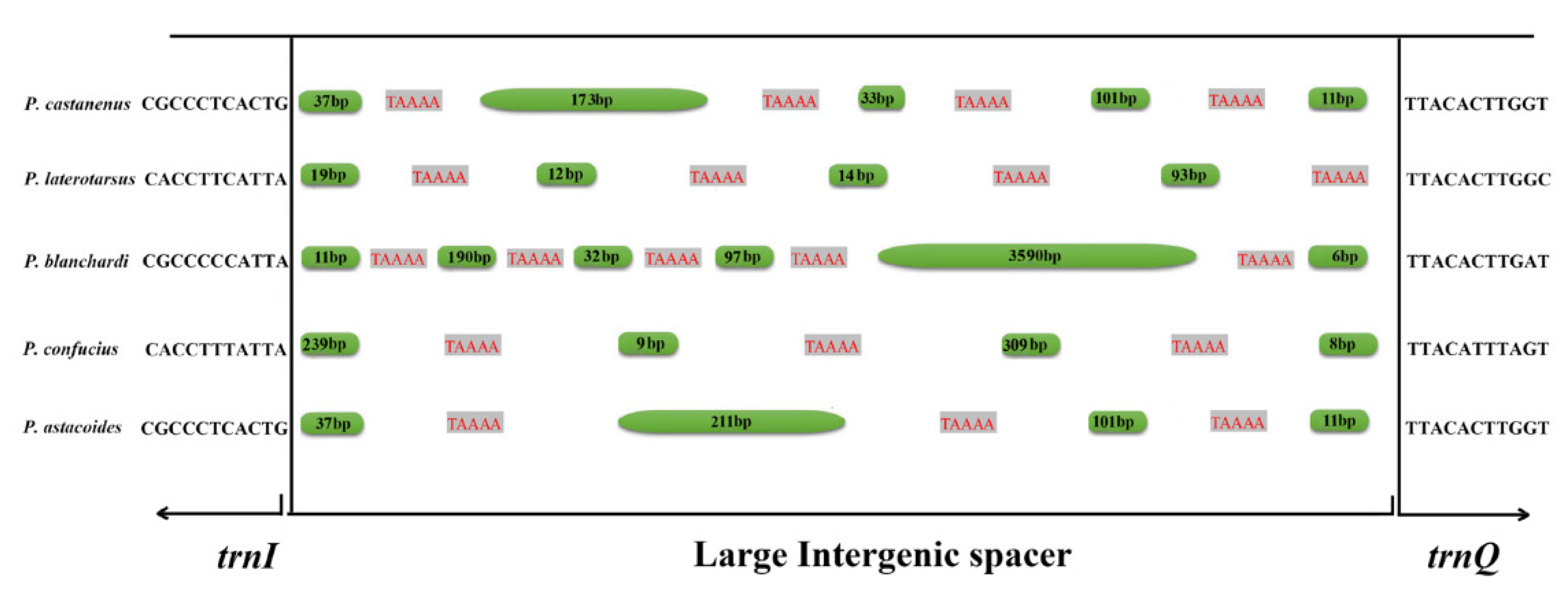
3.4. Intergenic Spacers
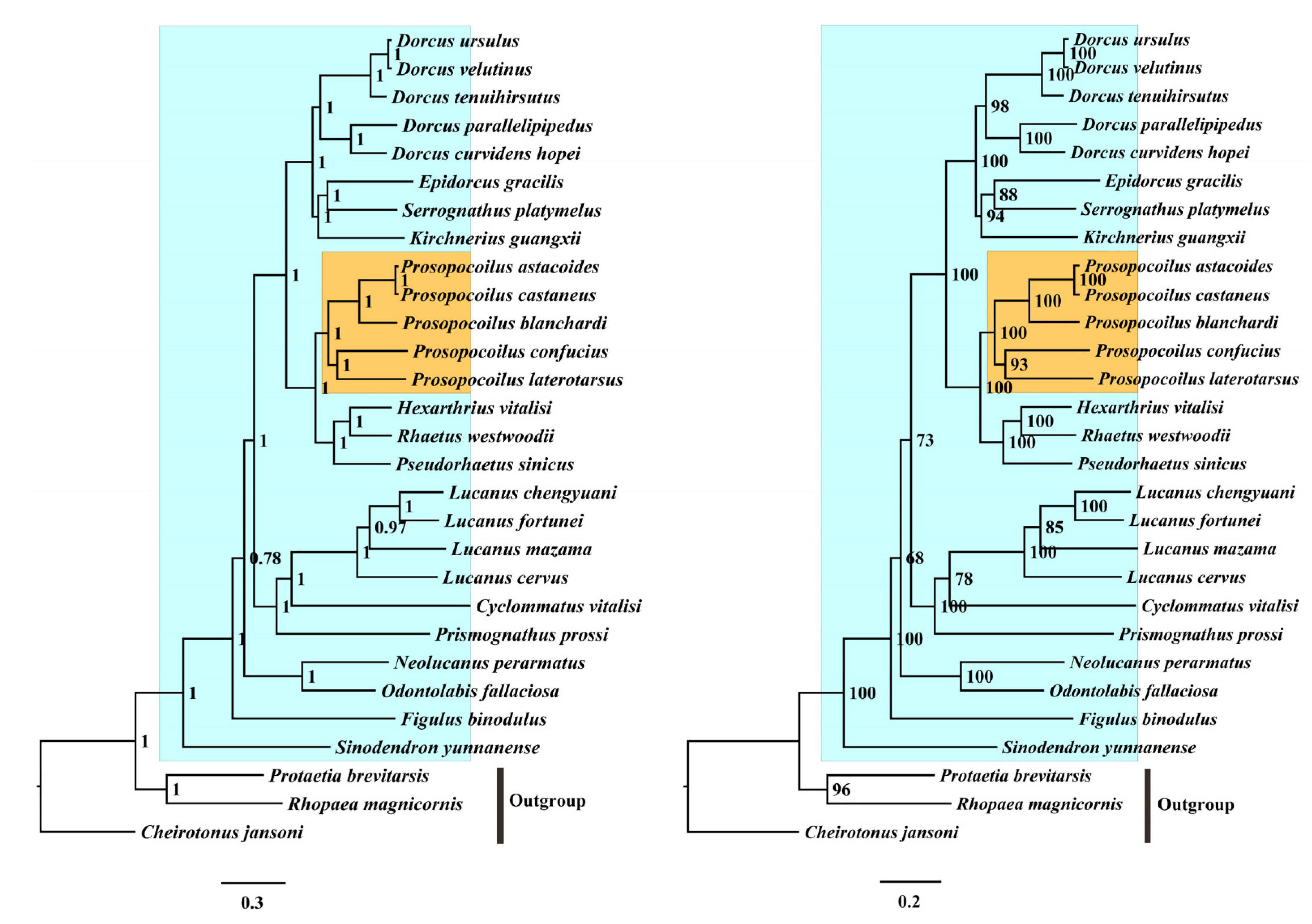
3.5. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, S.I.; Farrell, B.D. Phylogeny of world stag beetles (Coleoptera: Lucanidae) reveals a Gondwanan origin of Darwin’s stag beetle. Mol. Phylogenet. Evol. 2015, 86, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Havemann, N.; Gossner, M.M.; Hendrich, L.; Jèrôme, M.; Niedringhaus, R.; Peter, S.; Raupach, M.J. From water striders to water bugs: The molecular diversity of aquatic Heteroptera (Gerromorpha, Nepomorpha) of Germany based on DNA barcodes. PeerJ 2018, 6, e4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.Q.; Song, F.; Li, T.; Wu, Y.Y.; Wan, X. New mitogenomes of two Chinese stag beetles (Coleoptera, Lucanidae) and their implications for systematics. J. Insect Sci. 2017, 17, 63. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Vogler, A.P. Evolution: Taking the sting out of wasp phylogenetics. Curr. Biol. 2017, 27, R358–R360. [Google Scholar] [CrossRef]
- Timmermans, M.J.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Dodsworth, S.; Foster, P.G.; Bocak, L.; Vogler, A.P. Family-level sampling of mitochondrial genomes in Coleoptera: Compositional heterogeneity and phylogenetics. Genome Biol. Evol. 2016, 8, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Cong, Q.; Grishin, N.V. The complete mitochondrial genome of Lerema accius and its phylogenetic implications. PeerJ 2016, 4, e1546. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Zhang, L.; Lu, T.; Ma, J.; Zeng, C.; Yue, B.; Zhang, X.Y. Mitochondrial genomes of blister beetles (Coleoptera, Meloidae) and two large intergenic spacers in Hycleus genera. BMC Genom. 2017, 18, 698. [Google Scholar] [CrossRef]
- Zhang, L.P.; Cai, Y.Y.; Yu, D.N.; Storey, K.B.; Zhang, J.Y. Gene characteristics of the complete mitochondrial genomes of Paratoxodera polyacantha and Toxodera hauseri (Mantodea: Toxoderidae). PeerJ 2018, 6, e4595. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Leavengood, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.P.; Zhou, X.G.; Cai, W.Z. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. Biol. Sci. 2017, 284, 20171223. [Google Scholar] [CrossRef]
- Nie, R.E.; Breeschoten, T.; Timmermans, M.; Nadein, K.; Xue, H.J.; Bai, M.; Huang, Y.; Yang, X.K.; Vogler, A.P. The phylogeny of Galerucinae (Coleoptera: Chrysomelidae) and the performance of mitochondrial genomes in phylogenetic inference compared to nuclear rRNA genes. Cladistics–Int. J. Willi Hennig Soc. 2017, 34, 113–130. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.J.; Yang, D.X.; Xu, K.K.; Cao, Y.; Li, C. Complete mitochondrial genome of the bamboo snout beetle, Cyrotrachelus buqueti (Coleoptera: Curculionidae). Mitochondrial DNA Part B 2018, 3, 88–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; He, S.L.; Song, X.H.; Liao, Q.; Zhang, X.Y.; Yue, B.S. The complete mitochondrial genome of Epicauta chinensis (Coleoptera: Meloidae) and phylogenetic analysis among coleopteran insects. Gene 2016, 578, 274–280. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol. Biol. Evol. 2008, 25, 2499–2509. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Zhang, Q.; Zhang, L. High-level phylogeny of the Coleoptera inferred with mitochondrial genome sequences. Mol. Phylogenet. Evol. 2016, 104, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dai, X.Y.; Xu, X.D.; Zhang, Z.Y.; Yu, D.N.; Storey, K.B.; Zhang, J.Y. The complete mitochondrial genomes of five longicorn beetles (Coleoptera: Cerambycidae) and phylogenetic relationships within Cerambycidae. PeerJ 2019, 7, e7633. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.S.; Min, J.K.; Kim, I. The mitochondrial genome of the dung beetle, Copris tripartitus, with mitogenomic comparisons within Scarabaeidae (Coleoptera). Int. J. Biol. Macromol. 2020, 144, 874–891. [Google Scholar] [CrossRef]
- Zhang, S.; Sekerka, L.; Liao, C.; Long, C.; Xu, J.; Dai, X.; Guo, Q. The First Eight Mitogenomes of Leaf-Mining Dactylispa Beetles (Coleoptera: Chrysomelidae: Cassidinae) Shed New Light on Subgenus Relationships. Insects 2021, 12, 1005. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Kim, S.; Wan, X. Mitochondrial genomes of the Dorcus velutinus complex (Coleoptera: Lucanidae) with the large intergenic spacer showing unique short sequence repeats and their implications for systematics. J. Asia-Pac. Entomol. 2021, 24, 493–501. [Google Scholar] [CrossRef]
- Xiao, L.F.; Zhang, S.D.; Long, C.P.; Guo, Q.Y.; Xu, J.S.; Dai, X.H.; Wang, J.G. Complete Mitogenome of a Leaf-Mining Buprestid Beetle, Trachys auricollis, and Its Phylogenetic Implications. Genes 2019, 10, 992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, N.; Li, X.X.; Yin, X.M.; Xi, Y.Q. The mitochondrial genomes of ladybird beetles and implications for evolution and phylogeny. Int. J. Biol. Macromol. 2020, 147, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, K.G.; Kim, S.R.; Kim, I. Complete mitochondrial genome of the two-spotted stag beetle, Metopodontus blanchardi (Coleoptera: Lucanidae). Mitochondrial DNA 2013, 26, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, N.; Zhang, H. Comparative and phylogenomic analyses of mitochondrial genomes in Coccinellidae (Coleoptera: Coccinelloidea). PeerJ 2021, 9, e12169. [Google Scholar] [CrossRef]
- Xiao, J.; Liu, J.; Ma, L.; Hao, X.; Yu, R.; Yuan, X. Mitogenomes of Nine Asian Skipper Genera and Their Phylogenetic Position (Lepidoptera: Hesperiidae: Pyrginae). Insects 2022, 13, 68. [Google Scholar] [CrossRef]
- Wan, X. Study on the Systematics of Lucanidae from China (Coleoptera: Scarabaeoidea). Doctoral Dissertation, Institute of Zoology, Chinese Academy of Sciences, Beijing, China, 2007; pp. 1–421. [Google Scholar]
- Huang, H.; Chen, C.C. Stag Beetles of China II; Formosa Ecological Company: Taiwan, China, 2013; pp. 1–716. [Google Scholar]
- Arrow, G.J. Coleoptera, Lamellicornia, Lucanidae and Passalidae: The Fauna of India, including Pakistan, Ceylon, Burma and Malaya; Taylor and Francis: London, UK, 1950; pp. 1–275. [Google Scholar]
- Bartolozzi, L.; Sprecher-Uebersax, E. Lucanidae. In Catalogue of Palaearctic Coleoptera, Scarabaeoidea–Scirtoidea–Dascilloidea–Buprestoidea–Byrrhoidea; Apollo Books: Vester Skerninge, Denmark, 2006; pp. 63–76. [Google Scholar]
- Benesh, B. Lucanidae Coleopterorum Cataloguss Supplementa; Wilhelm Junk: Gravenhage, The Netherlands, 1960; Volume 8, pp. 1–178. [Google Scholar]
- Didier, R.; Séguy, E. Catalogue illustré des Lucanides du Globe. In Texte Encyclopé die Entomologique Série A; Paul Lechevalier: Paris, France, 1953; pp. 1–223. [Google Scholar]
- Fujita, H. The Lucanid Beetles of the World; Mushi-sha: Tokyo, Japan, 2010; pp. 1–472. [Google Scholar]
- Krajčík, M. Lucanidae of the world. In Catalogue-Part I, Checklist of the Stag Beetles of the World (Coleoptera: Lucanidae); Stampata in Proprio: Most, Czech Republic, 2001; pp. 1–108. [Google Scholar]
- Krajčík, M. Lucanidae of the world. In Catalogue-Part II, Encyclopaedia of the Lucanidae (Coleoptera: Lucanidae); Krajčík: Plzen, Czech Republic, 2008; pp. 1–197. [Google Scholar]
- Hosoya, T.; Araya, K. Phylogeny of Japanese stag beetles (Coleoptera: Lucanidae) inferred from 16S mtrRNA gene sequences, with reference to the evolution of sexual dimorphism of mandibles. Zool. Sci. 2005, 22, 1305–1318. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Chen, Y.J.; Ying, Y.; Yuan, J.J. The first complete mitogenome of skin beetles Omorgus chinensis (Coleoptera: Trogidae) with the phylogenetic implications. Mitochondrial DNA Part B 2022, 7, 70–73. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNA scan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Linbrado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.H.; Du, L.M.; Yue, B.S. An application for sequence retrieval and extraction from the GenBank flatfile. J. Hered. 2012, 103, 908–911. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [Green Version]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; Von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: A community resource for phylogenetic analyses. ACM 2011, 41, 1–8. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.G.; Liu, J.P.; Sun, C.H.; Vogler, A.P.; Cai, W.Z. Capturing the phylogeny of holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef]
- Chen, D.; Liu, J.; Bartolozzi, L.; Wan, X. The complete mitochondrial genome of stag beetle Lucanus cervus (Coleoptera: Lucanidae) and phylogenetic analysis. PeerJ 2019, 7, e8247. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Huang, J.P.; Shiao, S.F.; Ko, H.P.; Sung, C.H. Characterisation of the complete mitochondrial genome of Lucanus chengyuani (Coleoptera: Lucanidae). Mitochondrial DNA Part B 2019, 4, 3460–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Park, J.; Xi, H.; Park, J. Comprehensive Analyses of the Complete Mitochondrial Genome of Figulus binodulus (Coleoptera: Lucanidae). J. Insect Sci. 2020, 20, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.N.; Zhou, S.J.; Chen, Y.J.; Wan, X. The mitochondrial genome of a rare Chinese stag beetle Kirchnerius guangxii (Coleoptera: Lucanidae). Mitochondrial DNA Part B 2020, 5, 1633–1635. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.B.; Wang, J.; Wu, L.; Bai, Y.; Wang, C.; Qi, G.L.; Li, C.; Cao, Y. Characterization of the complete mitochondrial genome of Pseudorhaetus sinicus Boileau, 1899 (Coleoptera: Lucanidae). Mitochondrial DNA Part B 2021, 6, 3398–3399. [Google Scholar] [CrossRef]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef]
- Hua, J.M.; Li, M.; Dong, P.Z.; Cui, Y.; Xie, Q.; Bu, W.J. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genom. 2008, 9, 610. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.N.; Zhu, B.J.; Dai, L.S.; Wang, L.; Qian, C.; Wei, G.Q.; Liu, C.L. The complete mitochondrial genome of the common cutworm, Spodoptera litura (Lepidoptera: Noctuidade). Mitochondrial DNA Part A 2014, 27, 122–123. [Google Scholar] [CrossRef]
- Nardi, F.; Carapelli, A.; Fanciulli, P.P.; Dallai, R.; Frati, F. The complete mitochondrial DNA sequence of the basal hexapod Tetrodontophora bielanensis: Evidence for Heteroplasmy and tRNA translocations. Mol. Biol. Evol. 2001, 18, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Fenn, J.D.; Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome sequence of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Mol. Biol. 2010, 16, 239–252. [Google Scholar] [CrossRef]
- Kim, M.J.; Wang, A.R.; Park, J.S.; Kim, I. Complete mitochondrial genomes of five skippers (Lepidoptera: Hesperiidae) and phylogenetic reconstruction of Lepidoptera. Gene 2014, 549, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.Y.; Shi, Q.Y.; Ling, Y.; Chen, J.Y.; Zhang, B.F.; Li, X.J. Comparative Analysis of Mitogenomes among Five Species of Filchnerella (Orthoptera: Acridoidea: Pamphagidae) and Their Phylogenetic and Taxonomic Implications. Insects 2021, 12, 605. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Liu, J.; Cao, Y.Y.; Zhou, S.J.; Wan, X. Two new complete mitochondrial genomes of Dorcus stag beetles (Coleoptera, Lucanidae). Genes Genom. 2018, 40, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Schenk, K.D. Description of a new genus, two new taxa and one new subspecies of the family stag beetles from China, province Guangxi (Coleoptera, Lucanidae). Beetles World 2009, 2, 1–6. [Google Scholar]
- Schenk, K.D. Taxonomic notes to the family Lucanidae (Coleoptera, Lucanidae). Beetles World 2012, 6, 9–15. [Google Scholar]
- Huang, H.; Chen, C.C. Notes on Prosopocoilus Hope from China, with the description of two new species. Zootaxa 2011, 3126, 39–54. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Name | Sequence (5′-3′) | Length (bp) | Reference |
|---|---|---|---|---|
| cox1 | COI-F1 | CAACATTTATTTTGATTTTTTGG | 23 | [4] |
| COI-R1 | TCCAATGCACTAATCTGCCATATTA | 25 | [4] | |
| cytb | Cytb-F2 | GAGGAGCAACTGTAATTACTAA | 22 | [4] |
| Cytb-R2 | AAAAGAAARTATCATTCAGGTTGAAT | 26 | [4] | |
| 16S | 16S-F1 | CCGGTTTGAACTCAGATCATG | 21 | [4] |
| 16S-R1 | TAATTTATTGTACCTTGTGTATCAG | 25 | [4] |
| Family | Taxa | Length (bp) | I-Q/S2-1 | ACC. Number | References |
|---|---|---|---|---|---|
| Lucanidae | Cyclommatus vitalisi (Pouillaude, 1913) | 17,795 | S2-1 | MF037205 | [20] |
| (Ingroup) | Dorcus curvidens hopei (Nomura, 1960) | 16,026 | S2-1 | MF612067 | [20] |
| Dorcus parallelipipedus (Linnaeus, 1758) | 17,563 | S2-1 | JX412841 | [20] | |
| Dorcus tenuihirsutus (Kim & Kim, 2010) | 18,266 | S2-1 | MK050991 | [20] | |
| Dorcus ursulus (Arrow, 1938) | 18,001 | S2-1 | MK050990 | [20] | |
| Dorcus velutinus (Thomson, 1862) | 16,939 | S2-1 | MK050989 | [20] | |
| Epidorcus gracilis (Séguy, 1954) | 16,736 | S2-1 | KP735805 | [4] | |
| Lucanus cervus (Linnaeus, 1758) | 20,109 | S2-1 | MN580549 | [50] | |
| Lucanus fortunei (Saunders, 1854) | 16,591 | S2-1 | JX313688 | Unpublished | |
| Lucanus mazama (LeConte, 1861) | 15,261 | S2-1 | FJ613419 | [50] | |
| Lucanus chengyuani (Wang and Ko, 2018) | 16,926 | S2-1 | MK878514 | [51] | |
| Figulus binodulus (Waterhouse, 1873) | 16,261 | S2-1 | NC045102 | [52] | |
| Kirchnerius guangxii (Schenk, 2009) | 15,296 | S2-1 | MK134567 | [53] | |
| Neolucanus perarmatus (Didier, 1925) | 16,610 | S2-1 | MF401425 | Unpublished | |
| Odontolabis fallaciosa (Boileau, 1901) | 20,276 | S2-1 | MF908524 | [50] | |
| Prismognathus prossi (Bartolozzi & Wan, 2006) | 15,984 | S2-1 | MF614014 | [50] | |
| Prosopocoilus blanchardi (Parry, 1873) | 21,628 | I-Q/S2-1 | KF364622 | [23] | |
| Prosopocoilus castaneus (Hope et Westwood, 1845) | 17,523 | I-Q/S2-1 | ON401054 | This study | |
| Prosopocoilus confucius (Hope, 1842) | 16,951 | I-Q/S2-1 | KU552119 | [4] | |
| Prosopocoilus laterotarsus (Houlbert, 1915) | 17,333 | I-Q/S2-1 | ON401055 | This study | |
| Prosopocoilus astacoides (Hope, 1840) | 17,746 | I-Q/S2-1 | NC050851 | Unpublished | |
| Pseudorhaetus sinicus (Boileau, 1899) | 18,126 | S2-1 | MZ504793 | [54] | |
| Hexarthrius vitalisi (Didier, 1925) | 18,362 | S2-1 | JX313676 | Unpublished | |
| Rhaetus westwoodi (Parry, 1862) | 18,131 | S2-1 | MG159815 | [50] | |
| Serrognathus platymelus (Saunders, 1854) | 17,088 | S2-1 | MF612070 | Unpublished | |
| Sinodendron yunnanense (Král, 1994) | 16,921 | S2-1 | KP735804 | [4] | |
| Scarabaeidae | Cheirotonus jansoni (Jordan, 1898) | 17,249 | S2-1 | KC428100 | [4] |
| (Outgroup) | Protaetia brevitarsis (Lewis, 1879) | 20,319 | S2-1 | KC775706 | [20] |
| Rhopaea magnicornis (Blackburn, 1888) | 17,522 | S2-1 | FJ859903 | [50] |
| Gene | Strand | Region | Length (bp) | Start/Stop Codon | Intergenic (bp) |
|---|---|---|---|---|---|
| Pc/Pl | Pc/Pl | Pc/Pl | Pc/Pl | ||
| trnI | J | 1-64/335-398 | 64/64 | - | 375/158 |
| trnQ | N | 440-508/557-625 | 69/69 | - | −1/−1 |
| trnM | J | 508-575/625-693 | 68/69 | - | 0/0 |
| nad2 | J | 576-1589/694-1707 | 1014/1014 | ATA(TAA)/ATA(TAG) | 2/2 |
| trnW | J | 1592-1657/1710-1773 | 66/64 | - | −8/−8 |
| trnC | N | 1650-1710/1766-1825 | 61/60 | - | 0/0 |
| trnY | N | 1711-1775/1826-1889 | 65/64 | - | 1/1 |
| cox1 | J | 1777-3307/1891-3421 | 1531/1531 | AAC(T)/AAT(T) | 0/0 |
| trnL2 (UUR) | J | 3308-3372/3422-3485 | 65/64 | - | 0/0 |
| cox2 | J | 3373-4060/3486-4170 | 688/685 | ATA(T)/ATC(T) | 0/0 |
| trnK | J | 4061-4130/4171-4240 | 70/70 | - | 0/0 |
| trnD | J | 4131-4193/4241-4303 | 63/63 | - | 0/0 |
| atp8 | J | 4194-4349/4304-4459 | 156/156 | ATT(TAA)/ATT(TAA) | −4/−4 |
| atp6 | J | 4346-5014/4456-5124 | 669/669 | ATA(TAA)/ATA(TAA) | −1/−1 |
| cox3 | J | 5014-5797/5124-5907 | 784/784 | ATG(T)/ATG(T) | 0/0 |
| trnG | J | 5798-5861/5908-5970 | 64/63 | - | 0/0 |
| nad3 | J | 5862-6213/5971-6322 | 352/352 | ATG(T)/ATA(T) | 0/0 |
| trnA | J | 6214-6278/6323-6387 | 65/65 | - | −1/−1 |
| trnR | J | 6278-6341/6387-6450 | 64/64 | - | −1/−1 |
| trnN | J | 6341-6404/6450-6513 | 64/64 | - | 0/0 |
| trnS1 (AGN) | J | 6405-6471/6514-6580 | 67/67 | - | 0/0 |
| trnE | J | 6472-6535/6581-6644 | 64/64 | - | −2/−2 |
| trnF | N | 6534-6598/6643-6706 | 65/64 | - | 0/0 |
| nad5 | N | 6599-8315/6707-8423 | 1717/1717 | ATA(T)/ATT(T) | 0/0 |
| trnH | N | 8316-8379/8424-8487 | 64/64 | - | 0/0 |
| nad4 | N | 8380-9715/8488-9823 | 1336/1336 | ATG(T)/ATG(T) | −7/−7 |
| nad4l | N | 9709-9996/9817-10104 | 288/288 | ATG(TAA)/ATG(TAA) | 2/2 |
| trnT | J | 9999-10061/10107-10171 | 63/65 | - | 0/−1 |
| trnP | N | 10062-10127/10171-10236 | 66/66 | - | 5/5 |
| nad6 | J | 10133-10630/10242-10739 | 498/498 | ATG(TAA)/ATG(TAA) | −1/−1 |
| cytb | J | 10630-11770/10739-11879 | 1141/1141 | ATG(T)/ATG(T) | 0/0 |
| trnS2 (UCN) | J | 11771-11835/11880-11944 | 65/65 | - | 18/18 |
| nad1 | N | 11854-12804/11963-12913 | 951/951 | ATA(TAG)/ATA(TAG) | 0/0 |
| trnL1 (CUN) | N | 12805-12868/12914-12976 | 64/63 | - | 0/0 |
| rrnL | N | 12869-14136/12977-14242 | 1268/1266 | - | 0/0 |
| trnV | N | 14137-14205/14243-14311 | 69/69 | - | −1/0 |
| rrnS | N | 14205-15014/14312-15116 | 810/805 | - | 0/− |
| Control region | - | 15015-17523/- | 2509/- | - | 0/− |
| Species | Genes | T(U) | C | A | G | A + T% | G + C% | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|---|
| P. castaneus | PCGs | 39.14 | 17.06 | 28.19 | 15.61 | 67.32 | 32.68 | −0.16 | −0.04 |
| tRNAs | 33.45 | 13.66 | 36.66 | 16.24 | 70.10 | 29.90 | 0.05 | 0.09 | |
| rRNAs | 39.03 | 8.86 | 31.42 | 20.70 | 70.44 | 29.56 | −0.11 | 0.40 | |
| Genome | 32.07 | 20.33 | 36.67 | 10.93 | 68.73 | 31.27 | 0.07 | −0.30 | |
| P. laterotarsus | PCGs | 40.00 | 16.49 | 27.64 | 15.88 | 67.63 | 32.37 | −0.18 | −0.02 |
| tRNAs | 34.20 | 12.31 | 37.55 | 15.94 | 71.75 | 28.25 | 0.05 | 0.13 | |
| rRNAs | 38.77 | 8.64 | 32.83 | 19.75 | 71.61 | 28.39 | −0.08 | 0.39 | |
| Genome | 32.81 | 19.82 | 36.44 | 10.93 | 69.26 | 30.74 | 0.05 | −0.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, M.; Zhou, S.; Wan, X. Phylogenetic Implication of Large Intergenic Spacers: Insights from a Mitogenomic Comparison of Prosopocoilus Stag Beetles (Coleoptera: Lucanidae). Animals 2022, 12, 1595. https://doi.org/10.3390/ani12131595
Xu M, Zhou S, Wan X. Phylogenetic Implication of Large Intergenic Spacers: Insights from a Mitogenomic Comparison of Prosopocoilus Stag Beetles (Coleoptera: Lucanidae). Animals. 2022; 12(13):1595. https://doi.org/10.3390/ani12131595
Chicago/Turabian StyleXu, Mengqiong, Shiju Zhou, and Xia Wan. 2022. "Phylogenetic Implication of Large Intergenic Spacers: Insights from a Mitogenomic Comparison of Prosopocoilus Stag Beetles (Coleoptera: Lucanidae)" Animals 12, no. 13: 1595. https://doi.org/10.3390/ani12131595