
Periodical Changes of Feces Microbiota and Its Relationship with Nutrient Digestibility in Early Lambs

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experiment Design and Animal Management
2.2. Sample Collection and Measurement of Nutrient Digestion
2.3. Bacterial DNA Extraction
2.4. PCR Amplification and 16S rDNA Sequencing
2.5. Sequence and Statistical Analysis
3. Results
3.1. Microbial Diversity Analysis
3.1.1. Alpha Diversity
3.1.2. Comparison between Microbial Communities (Beta Diversity)
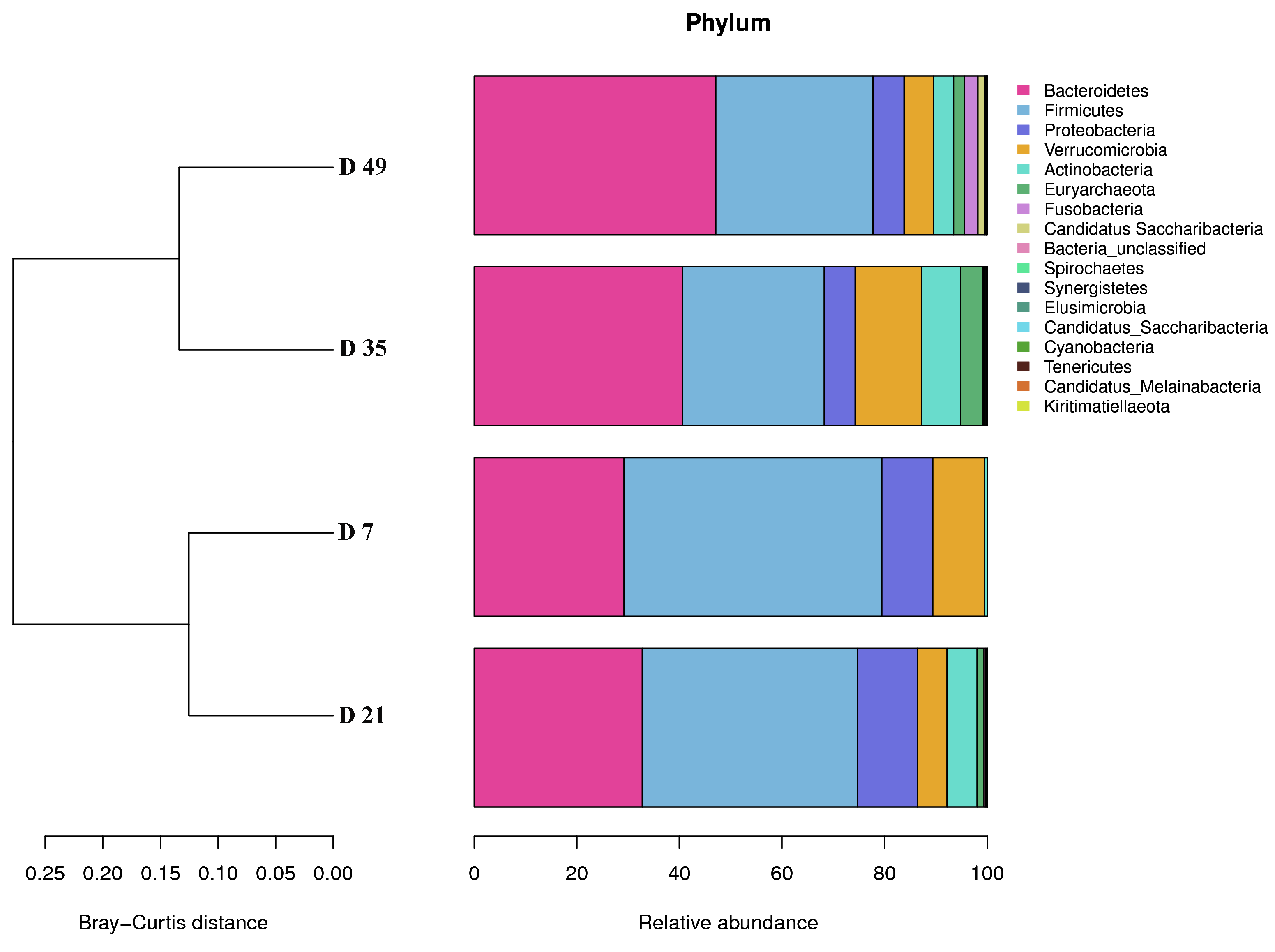
3.1.3. Phylogenetic Composition of Fecal Microorganism Communities
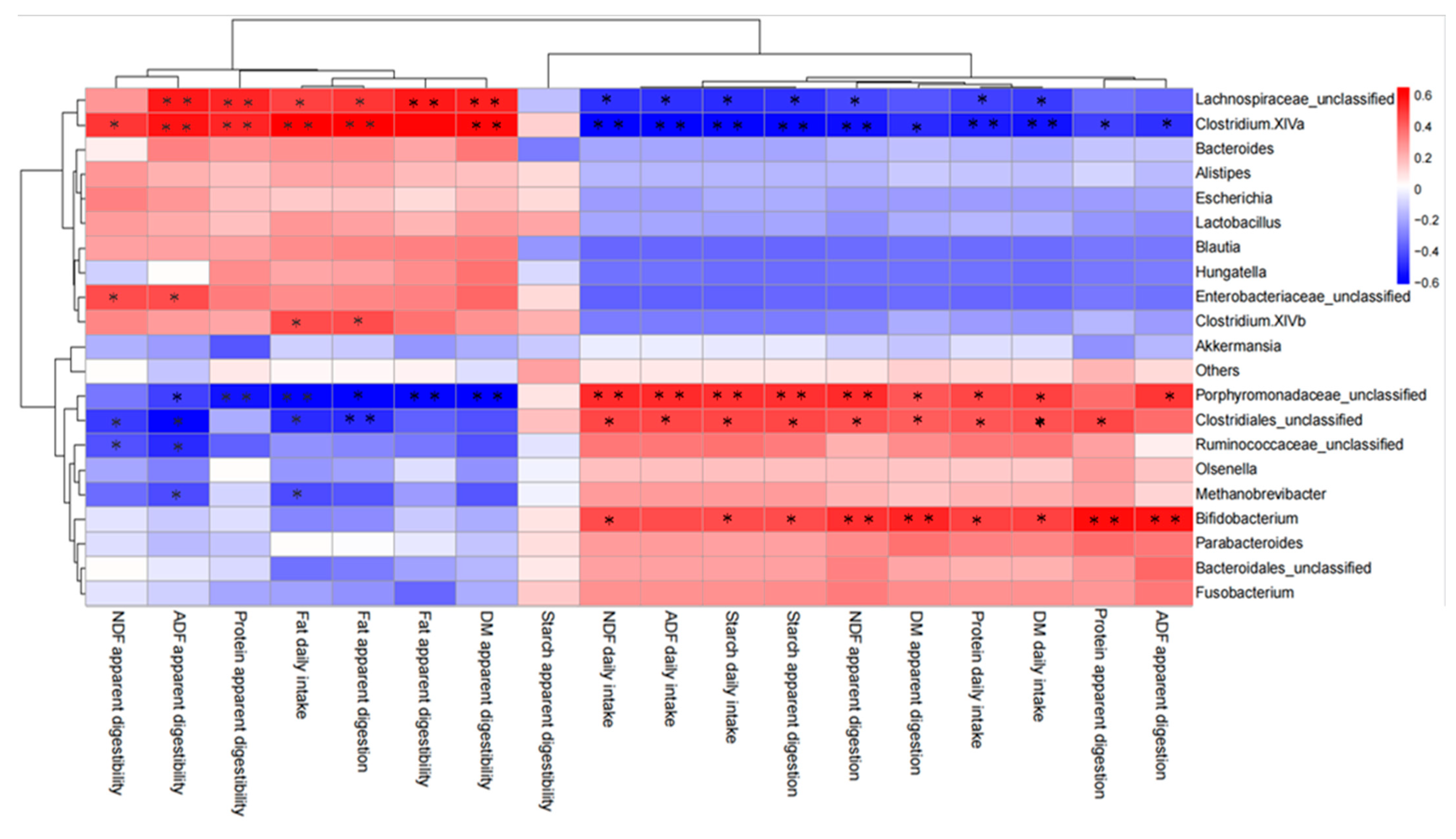
3.2. Dynamic Changes of Nutrient Digestibility in Lambs and Its Relationship with Fecal Microorganisms
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.P.; Dumas, M.E.; Wang, Y.; Legido-Quigley, C.; Yap, I.K.; Tang, H.; Zirah, S.; Murphy, G.M.; Cloarec, O.; Lindon, J.C.; et al. A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol. Syst. Biol. 2007, 3, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Holman, D.B.; Alexander, T.; Hu, T.; Wang, Y. Fecal microbiota of lambs fed purple prairie clover (Dalea purpurea Vent.) and alfalfa (Medicago sativa). Arch. Microbiol. 2017, 200, 137–145. [Google Scholar]
- Cholewińska, P.; Górniak, W.; Wojnarowski, K. Impact of selected environmental factors on microbiome of the digestive tract of ruminants. BMC Vet. Res. 2021, 17, 25. [Google Scholar] [CrossRef]
- Fonty, G.; Senaud, J.; Jouany, J.P.; Gouet, P. Establishment of ciliate protozoa in the rumen of conventional and conventionalized lambs: Influence of diet and management conditions. Can. J. Microbiol. 1988, 34, 235–241. [Google Scholar] [CrossRef]
- Yin, X.; Ji, S.; Duan, C.; Tian, P.; Ju, S.; Yan, H.; Zhang, Y.; Liu, Y. Age-Related Changes in the Ruminal Microbiota and Their Relationship With Rumen Fermentation in Lambs. Front. Microbiol. 2021, 12, 679135. [Google Scholar] [CrossRef]
- Furman, O.; Shenhav, L.; Sasson, G.; Kokou, F.; Honig, H.; Jacoby, S.; Hertz, T.; Cordero, O.X.; Halperin, E.; Mizrahi, I. Stochasticity constrained by deterministic effects of diet and age drive rumen microbiome assembly dynamics. Nat. Commun. 2020, 11, 1904. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wang, W.; Liu, T.; Zhang, Q.; Wang, G.; Li, F.; Li, F.; Yue, X.; Li, T. Effect of Early Weaning on the Intestinal Microbiota and Expression of Genes Related to Barrier Function in Lambs. Front. Microbiol. 2018, 9, 1431. [Google Scholar] [CrossRef] [Green Version]
- Bagath, M.; Krishnan, G.; Devaraj, C.; Rashamol, V.P.; Pragna, P.; Lees, A.M.; Sejian, V. The impact of heat stress on the immune system in dairy cattle: A review. Res. Vet. Sci. 2019, 126, 94–102. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Q.; Wang, G.; Niu, X.; Wang, W.; Li, F.; Li, F.; Zhang, Z. The functional development of the rumen is influenced by weaning and associated with ruminal microbiota in lambs. Anim. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, G.; Zhang, Q.; Huang, Y.; Li, F.; Wang, W. Developmental changes of nutrient digestion in young lambs are influenced by weaning and associated with intestinal microbiota. Anim. Biotechnol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Xie, F.; Sun, D.; Liu, J.; Zhu, W.; Mao, S. Ruminal microbiome-host crosstalk stimulates the development of the ruminal epithelium in a lamb model. Microbiome 2019, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, L.; Han, X.; Zhao, N.; Xu, T.; Ma, L.; Wang, X.; Zhang, X.; Kang, S.; Zhao, X.; et al. Tibetan Sheep Adapt to Plant Phenology in Alpine Meadows by Changing Rumen Microbial Community Structure and Function. Front. Microbiol. 2020, 11, 587558. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xu, T.; Xu, S.; Ma, L.; Han, X.; Wang, X.; Zhang, X.; Hu, L.; Zhao, N.; Chen, Y.; et al. Effect of dietary concentrate to forage ratio on growth performance, rumen fermentation and bacterial diversity of Tibetan sheep under barn feeding on the Qinghai-Tibetan plateau. PeerJ 2019, 7, e7462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharechahi, J.; Zahiri, H.S.; Noghabi, K.A.; Salekdeh, G.H. In-depth diversity analysis of the bacterial community resident in the camel rumen. Syst. Appl. Microbiol. 2015, 38, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.E.; Hume, I.D. Contributions of microbes in vertebrate gastrointestinal tract to production and conservation of nutrients. Physiol. Rev. 1998, 78, 393–427. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Guryn, K.; Leone, V.; Chang, E.B. Regional Diversity of the Gastrointestinal Microbiome. Cell Host Microbe 2019, 26, 314–324. [Google Scholar] [CrossRef]
- Xie, F.; Jin, W.; Si, H.; Yuan, Y.; Mao, S. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome 2021, 9, 137. [Google Scholar] [CrossRef]
- Mcdonald, P.; Edwards, R.A.; Greenhalgh, J.; Morgan, C.A. Animal Nutrition; Longman Scientific and Technical Inc.: New York, NY, USA, 1995. [Google Scholar]
- van Heugten, E.; Funderburke, D.W.; Dorton, K.L. Growth performance, nutrient digestibility, and fecal microflora in weanling pigs fed live yeast. J. Anim. Sci. 2003, 81, 1004–1012. [Google Scholar] [CrossRef]
- Van Soest, P.J.; Robertson, J.B.; Lewis, B.A. Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- Roswell, M.; Dushoff, J.; Winfree, R. A conceptual guide to measuring species diversity. Oikos 2021, 130, 321–338. [Google Scholar] [CrossRef]
- Chao, A.; Kubota, Y.; Zelen, D.; Chiu, C.; Colwell, R.K. Quantifying sample completeness and comparing diversities among assemblages. Ecol. Res. 2020, 35, 292–314. [Google Scholar] [CrossRef]
- Haegeman, B.; Hamelin, J.; Moriarty, J.; Neal, P.; Dushoff, J.; Weitz, J.S. Robust estimation of microbial diversity in theory and in practice. ISME J. 2013, 7, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, M.L.; Indugu, N.; Vecchiarelli, B.; Bender, J.; Pappalardo, C.; Leibstein, M.; Toth, J.; Katepalli, A.; Garapati, S.; Pitta, D. Temporal changes in the fecal bacterial community in Holstein dairy calves from birth through the transition to a solid diet. PLoS ONE 2020, 15, e0238882. [Google Scholar] [CrossRef]
- Gomez, D.E.; Arroyo, L.G.; Costa, M.C.; Viel, L.; Weese, J.S. Characterization of the Fecal Bacterial Microbiota of Healthy and Diarrheic Dairy Calves. J. Vet. Intern. Med. 2017, 31, 928–939. [Google Scholar] [CrossRef]
- Pitta, D.W.; Indugu, N.; Vecchiarelli, B.; Rico, D.E.; Harvatine, K.J. Alterations in ruminal bacterial populations at induction and recovery from diet-induced milk fat depression in dairy cows. J. Dairy Sci. 2018, 101, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Jewell, K.A.; McCormick, C.A.; Odt, C.L.; Weimer, P.J.; Suen, G. Ruminal Bacterial Community Composition in Dairy Cows Is Dynamic over the Course of Two Lactations and Correlates with Feed Efficiency. Appl. Environ. Microbiol. 2015, 81, 4697–4710. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.A.; Nachman, M.W. Spatial Heterogeneity of Gut Microbial Composition along the Gastrointestinal Tract in Natural Populations of House Mice. PLoS ONE 2016, 11, e0163720. [Google Scholar] [CrossRef] [Green Version]
- Sekelja, M.; Rud, I.; Knutsen, S.H.; Denstadli, V.; Westereng, B.; Næs, T.; Rudi, K. Abrupt temporal fluctuations in the chicken fecal microbiota are explained by its gastrointestinal origin. Appl. Environ. Microbiol. 2012, 78, 2941–2948. [Google Scholar] [CrossRef] [Green Version]
- Frutos, J.; Andrés, S.; Yáñez-Ruiz, D.R.; Benavides, J.; López, S.; Santos, A.; Martínez-Valladares, M.; Rozada, F.; Giráldez, F.J. Early feed restriction of lambs modifies ileal epimural microbiota and affects immunity parameters during the fattening period. Animal 2018, 12, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Doré, J.; Blottière, H. The influence of diet on the gut microbiota and its consequences for health. Curr. Opin. Biotechnol. 2015, 32, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [Green Version]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Bäckhed, F. Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Sanchez Ramos, L.; Rodloff, A.C. Identification of Clostridium species using the VITEK(®) MS. Anaerobe 2018, 54, 217–223. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Boutard, M.; Cerisy, T.; Nogue, P.Y.; Alberti, A.; Weissenbach, J.; Salanoubat, M.; Tolonen, A.C. Functional diversity of carbohydrate-active enzymes enabling a bacterium to ferment plant biomass. PLoS Genet. 2014, 10, e1004773. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Qi, N.; Zeng, Y.; Bao, M.; Chen, Y.; Liao, J.; Wei, L.; Cao, D.; Huang, S.; Luo, Q.; et al. The Endogenous Alterations of the Gut Microbiota and Feces Metabolites Alleviate Oxidative Damage in the Brain of LanCL1 Knockout Mice. Front. Microbiol. 2020, 11, 557342. [Google Scholar] [CrossRef]
- Burrows, C.F.; Kronfeld, D.S.; Banta, C.A.; Merritt, A.M. Effects of fiber on digestibility and transit time in dogs. J. Nutr. 1982, 112, 1726–1732. [Google Scholar] [CrossRef]
- El-Wahab, A.A.; Wilke, V.; Grone, R.; Visscher, C. Nutrient Digestibility of a Vegetarian Diet with or without the Supplementation of Feather Meal and Either Corn Meal, Fermented Rye or Rye and Its Effect on Fecal Quality in Dogs. Animals 2021, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Huhtanen, P.; Ahvenjärvi, S.; Broderick, G.A.; Reynal, S.M.; Shingfield, K.J. Quantifying ruminal digestion of organic matter and neutral detergent fiber using the omasal sampling technique in cattle--a meta-analysis. J. Dairy Sci. 2010, 93, 3203–3215. [Google Scholar] [CrossRef] [PubMed]
- Moharrery, A.; Larsen, M.; Weisbjerg, M.R. Starch digestion in the rumen, small intestine, and hind gut of dairy cows—A meta-analysis. Anim. Feed Sci. Technol. 2014, 192, 1–14. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Starter 1 | Milk Replacer |
|---|---|---|
| Ingredients [%] | ||
| Alfalfa meal | 18.50 | |
| Corn | 21.00 | |
| Extruded corn | 22.30 | |
| Bran | 6.00 | |
| Soybean meal | 21.50 | |
| Extruded soybean | 4.00 | |
| Corn gluten meal | 5.00 | |
| Limestone | 0.30 | |
| Premix 2 | 1.00 | |
| NaCl | 0.40 | |
| Total | 100.00 | |
| Chemical composition | ||
| DM (%) | 90.96 | 96.91 |
| DE (MJ·kg−1) | 13.01 | / |
| CP (%) | 19.50 | 23.22 |
| Fat (%) | 1.33 | 13.20 |
| Starch (%) | 33.10 | 0.00 |
| NDF (%) | 18.87 | 0.00 |
| ADF (%) | 8.60 | 0.00 |
| Diversity Indices | Groups | SME | p-Value | |||
|---|---|---|---|---|---|---|
| D7 | D21 | D35 | D49 | |||
| Observed index | 267.67 a | 446.67 b | 525.17 b | 526.00 b | 25.445 | <0.001 |
| Shannon index | 5.08 a | 6.18 b | 5.73 b | 5.72 ab | 0.135 | 0.026 |
| Simpson index | 0.94 | 0.96 | 0.93 | 0.95 | 0.008 | 0.376 |
| Chao1 index | 327.37 a | 579.17 b | 651.14 bc | 703.99 c | 33.452 | <0.001 |
| Genus | Groups | SEM | p-Value | |||
|---|---|---|---|---|---|---|
| D7 (%) | D21 (%) | D35 (%) | D49 (%) | |||
| Bacteroides | 27.26 | 18.42 | 9.94 | 19.63 | 2.252 | 0.101 |
| Porphyromonadaceae_unclassified | 0.34 a | 5.47 a | 22.06 b | 19.06 b | 2.956 | 0.018 |
| Lachnospiraceae_unclassified | 19.69 b | 12.37 ab | 6.46 a | 5.14 a | 2.074 | 0.034 |
| Akkermansia | 10.07 | 5.79 | 16.24 | 5.94 | 2.722 | 0.067 |
| Ruminococcaceae_unclassified | 3.39 | 5.32 | 3.11 | 10.57 | 1.241 | 0.076 |
| Clostridiales_unclassified | 2.32 | 1.81 | 7.28 | 5.84 | 0.891 | 0.100 |
| Escherichia | 2.73 | 5.09 | 0.72 | 3.08 | 0.970 | 0.582 |
| Enterobacteriaceae_unclassified | 5.74 | 2.61 | 0.24 | 0.51 | 1.087 | 0.268 |
| Clostridium XlVa | 5.23 b | 5.13 b | 0.41 a | 0.56 a | 0.740 | 0.009 |
| Blautia | 7.23 | 2.32 | 0.29 | 0.25 | 1.322 | 0.204 |
| Alistipes | 0.26 | 4.63 | 0.76 | 1.51 | 0.650 | 0.074 |
| Olsenella | 0.11 | 2.41 | 3.64 | 2.15 | 0.586 | 0.266 |
| Methanobrevibacter | 0.01 | 1.31 | 4.03 | 2.46 | 0.621 | 0.192 |
| Lactobacillus | 3.27 | 0.46 | 0.01 | 0.85 | 0.457 | 0.056 |
| Parabacteroides | 0.59 | 1.72 | 0.65 | 1.80 | 0.268 | 0.211 |
| Bacteroidales_unclassified | 0.01 | 0.53 | 6.23 | 1.22 | 0.903 | 0.128 |
| Bifidobacterium | 0.01 | 0.66 | 2.45 | 1.90 | 0.516 | 0.384 |
| Clostridium XlVb | 0.12 a | 3.83 b | 0.21 a | 0.35 a | 0.460 | 0.030 |
| Fusobacterium | 0.01 | 0 | 0.13 | 2.18 | 0.613 | 0.460 |
| Hungatella | 2.73 | 0.26 | 0.16 | 0.01 | 0.471 | 0.115 |
| Others | 8.89 | 19.87 | 14.99 | 14.99 | 1.585 | 0.101 |
| Item | Groups | SEM | p-Value | |||
|---|---|---|---|---|---|---|
| D7 | D21 | D35 | D49 | |||
| Milk replacer intake (kg/d) | 0.63 | 0.70 | / | / | / | / |
| Starter intake (kg/d) | 0.02 b | 0.06 b | 0.39 a | 0.49 a | 0.033 | <0.001 |
| Body weight (kg) | 4.63 c | 5.72 c | 7.74 b | 10.17 a | 0.572 | <0.001 |
| Average daily gain (kg/d) | 0.20 a | 0.08 b | 0.14 c | 0.17 cd | 0.024 | <0.001 |
| Item | Groups | SEM | p-Value | ||||
|---|---|---|---|---|---|---|---|
| D7 | D21 | D35 | D49 | ||||
| Protein | Apparent digestibility (%) | 86.53 b | 83.59 b | 66.50 a | 63.09 a | 2.630 | <0.001 |
| Daily intake (g/d) | 28.03 a | 39.40 a | 67.39 b | 94.84 c | 5.926 | <0.001 | |
| Apparent digestion (g/d) | 24.26 a | 32.81 ab | 45.53 b | 59.81 c | 3.464 | <0.001 | |
| Daily excretion (g/d) | 3.72 c | 6.58 c | 21.86 b | 35.04 a | 3.574 | <0.001 | |
| Starch | Apparent digestibility (%) | 84.99 | 88.65 | 86.95 | 87.33 | 0.695 | 0.329 |
| Daily intake (g/d) | 5.01 a | 22.40 a | 114.37 b | 160.96 c | 14.038 | <0.001 | |
| Apparent digestion (g/d) | 4.41 a | 19.91 a | 99.51 b | 140.82 c | 12.305 | <0.001 | |
| Daily excretion (g/d) | 0.60 c | 2.49 c | 14.86 b | 20.14 a | 1.946 | <0.001 | |
| Fat | Apparent digestibility (%) | 91.09 b | 90.66 b | 69.57 a | 66.87 a | 2.558 | <0.001 |
| Daily intake (g/d) | 14.46 c | 16.70 d | 4.47 a | 6.49 b | 1.088 | <0.001 | |
| Apparent digestion (g/d) | 13.17 c | 15.14 d | 3.13 a | 4.37 b | 1.108 | <0.001 | |
| Daily excretion (g/d) | 1.29 b | 1.56 b | 1.34 b | 2.13 a | 0.186 | <0.001 | |
| DM | Apparent digestibility (%) | 89.40 b | 86.27 b | 69.94 a | 68.78 a | 2.044 | <0.001 |
| Daily intake (g/d) | 118.87 a | 171.67 a | 324.36 b | 456.50 c | 29.997 | < 0.001 | |
| Apparent digestion (g/d) | 106.22 a | 147.58 a | 228.50 b | 315.14 c | 19.187 | <0.001 | |
| Daily excretion (g/d) | 12.65 c | 24.09 c | 95.86 b | 141.35 a | 9.770 | <0.001 | |
| NDF | Apparent digestibility (%) | 67.44 b | 73.93 b | 54.37 a | 53.03 a | 2.702 | 0.005 |
| Daily intake (g/d) | 2.87 a | 11.06 a | 65.44 b | 92.09 c | 8.113 | <0.001 | |
| Apparent digestion (g/d) | 2.23 a | 7.97 a | 36.13 b | 49.32 c | 4.463 | <0.001 | |
| Daily excretion (g/d) | 0.64 c | 3.09 c | 29.30 b | 42.77 a | 3.262 | <0.001 | |
| ADF | Apparent digestibility (%) | 69.45 b | 68.61 b | 38.03 a | 35.90 a | 4.165 | <0.001 |
| Daily intake (g/d) | 1.30 a | 5.02 a | 29.72 b | 41.83 c | 3.685 | <0.001 | |
| Apparent digestion (g/d) | 0.99 a | 3.29 a | 11.72 b | 15.39 b | 1.504 | <0.001 | |
| Daily excretion (g/d) | 12.01 c | 21.01 c | 66.56 b | 98.58 a | 7.142 | <0.001 | |
| Item (%) | Groups | SEM | p-Value | |||
|---|---|---|---|---|---|---|
| D7 | D21 | D35 | D49 | |||
| Fat | 1.87 a | 1.26 b | 0.98 b | 0.96 b | 0.121 | 0.016 |
| Dry matter | 69.53 a | 74.60 a | 60.50 b | 53.53 b | 2.102 | <0.001 |
| Protein | 33.80 a | 26.30 a | 14.35 b | 13.93 b | 2.132 | <0.001 |
| Acid detergent fiber | 4.57 a | 8.66 a | 13.63 b | 11.86 b | 0.877 | <0.001 |
| Neutral detergent fiber | 9.21 a | 14.54 b | 21.00 c | 18.06 bc | 1.161 | <0.001 |
| Starch | 4.05 | 4.05 | 4.38 | 3.15 | 0.499 | 0.857 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Wang, G.; Li, C.; Wang, W.; Zhang, X.; Wang, X.; Zhang, D.; Chen, Z.; Cui, P.; Ma, Z. Periodical Changes of Feces Microbiota and Its Relationship with Nutrient Digestibility in Early Lambs. Animals 2022, 12, 1770. https://doi.org/10.3390/ani12141770
Huang Y, Wang G, Li C, Wang W, Zhang X, Wang X, Zhang D, Chen Z, Cui P, Ma Z. Periodical Changes of Feces Microbiota and Its Relationship with Nutrient Digestibility in Early Lambs. Animals. 2022; 12(14):1770. https://doi.org/10.3390/ani12141770
Chicago/Turabian StyleHuang, Yongliang, Guoxiu Wang, Chong Li, Weimin Wang, Xiaoxue Zhang, Xiaojuan Wang, Deyin Zhang, Zhanyu Chen, Panpan Cui, and Zongwu Ma. 2022. "Periodical Changes of Feces Microbiota and Its Relationship with Nutrient Digestibility in Early Lambs" Animals 12, no. 14: 1770. https://doi.org/10.3390/ani12141770
APA StyleHuang, Y., Wang, G., Li, C., Wang, W., Zhang, X., Wang, X., Zhang, D., Chen, Z., Cui, P., & Ma, Z. (2022). Periodical Changes of Feces Microbiota and Its Relationship with Nutrient Digestibility in Early Lambs. Animals, 12(14), 1770. https://doi.org/10.3390/ani12141770