
Plateau Adaptation Gene Analyses Reveal Transcriptomic, Proteomic, and Dual Omics Expression in the Lung Tissues of Tibetan and Yorkshire Pigs

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal and Sample Preparation
2.3. Total RNA and Protein Isolation from Lung Samples
2.4. Library Preparation and RNA Sequencing
2.5. Differential Gene Analysis Using RNA-seq
2.6. Proteolysis and Labelling
2.7. Database Search, Protein Identification, and Quantification
2.8. RT-qPCR of Candidate Genes
3. Results
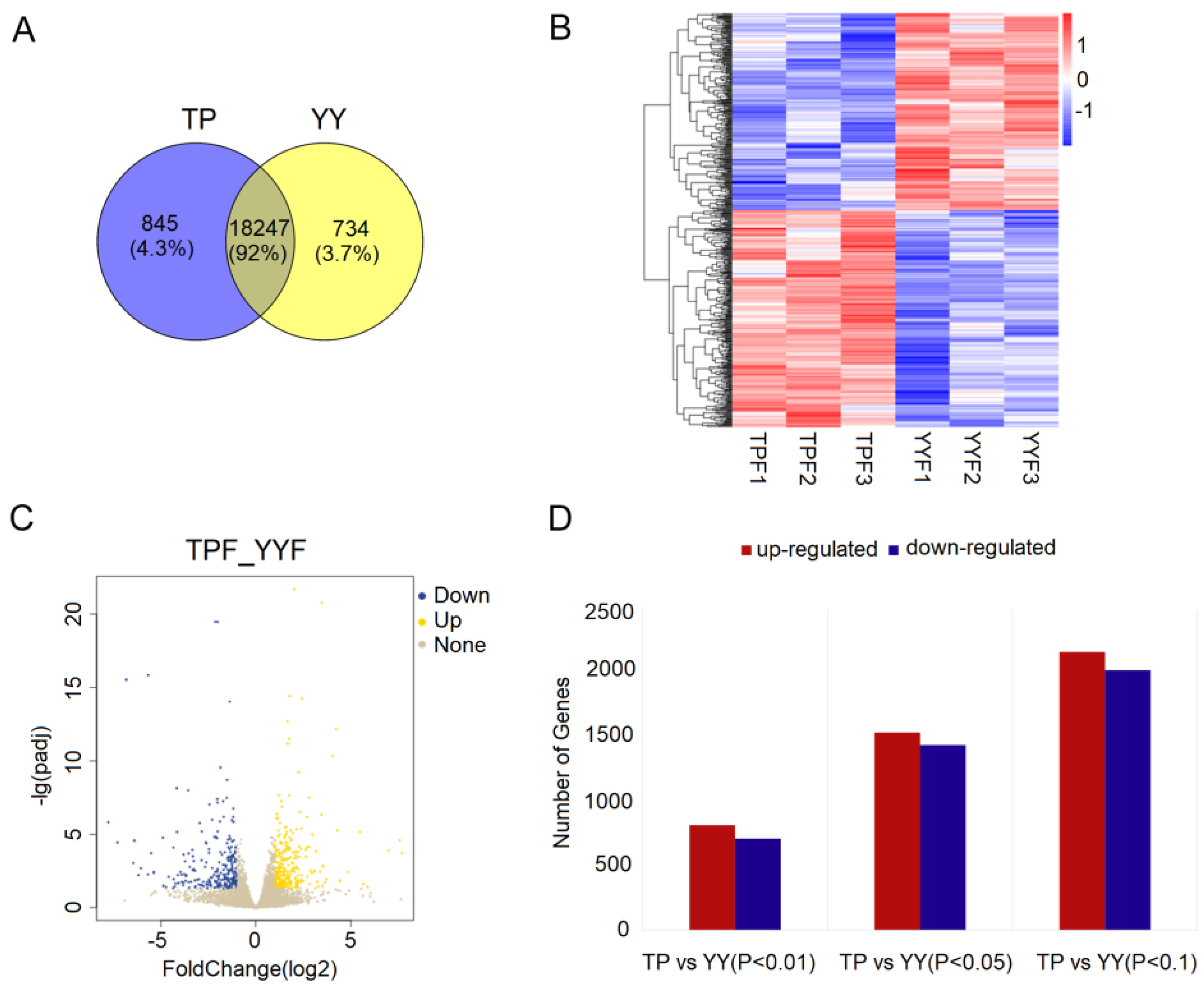
3.1. Summary of RNA-seq Data
3.2. Functional Annotation of DEGs
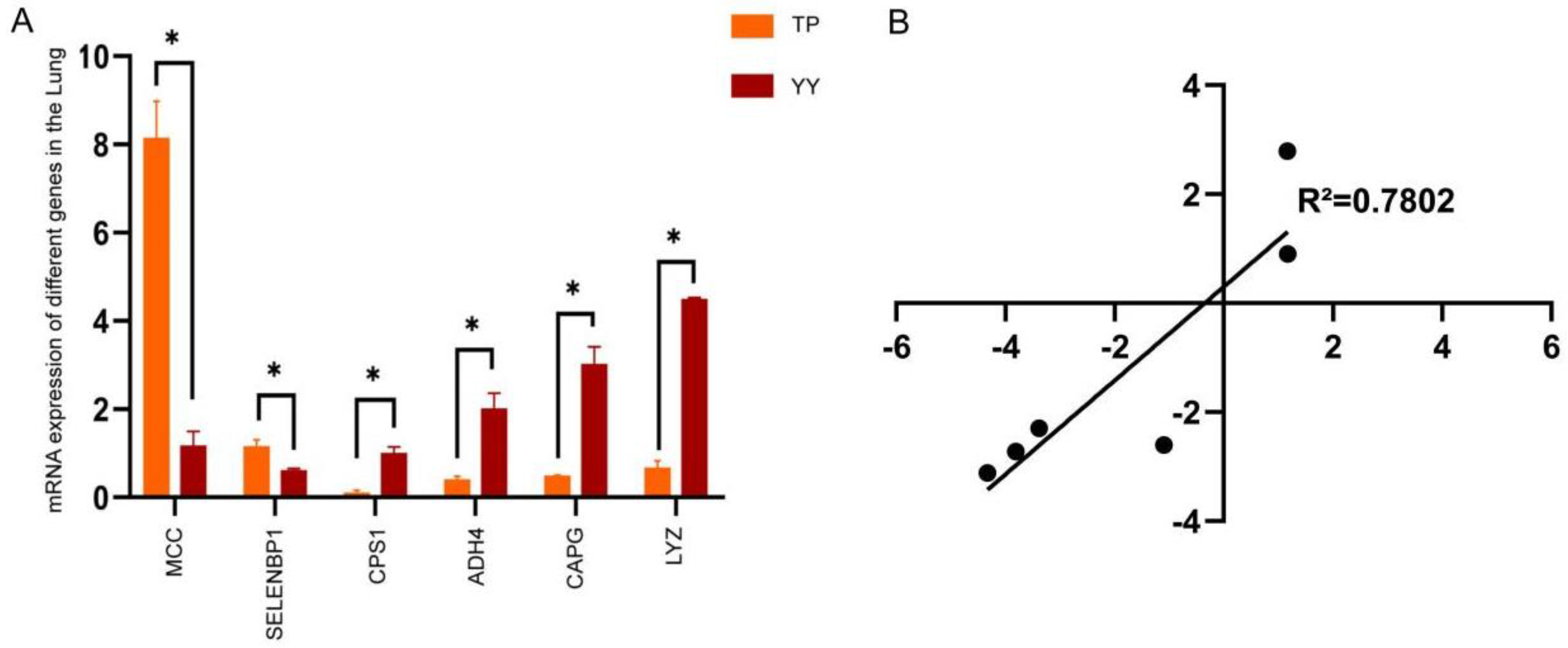
3.3. RNA-seq Date Validation by RT-qPCR
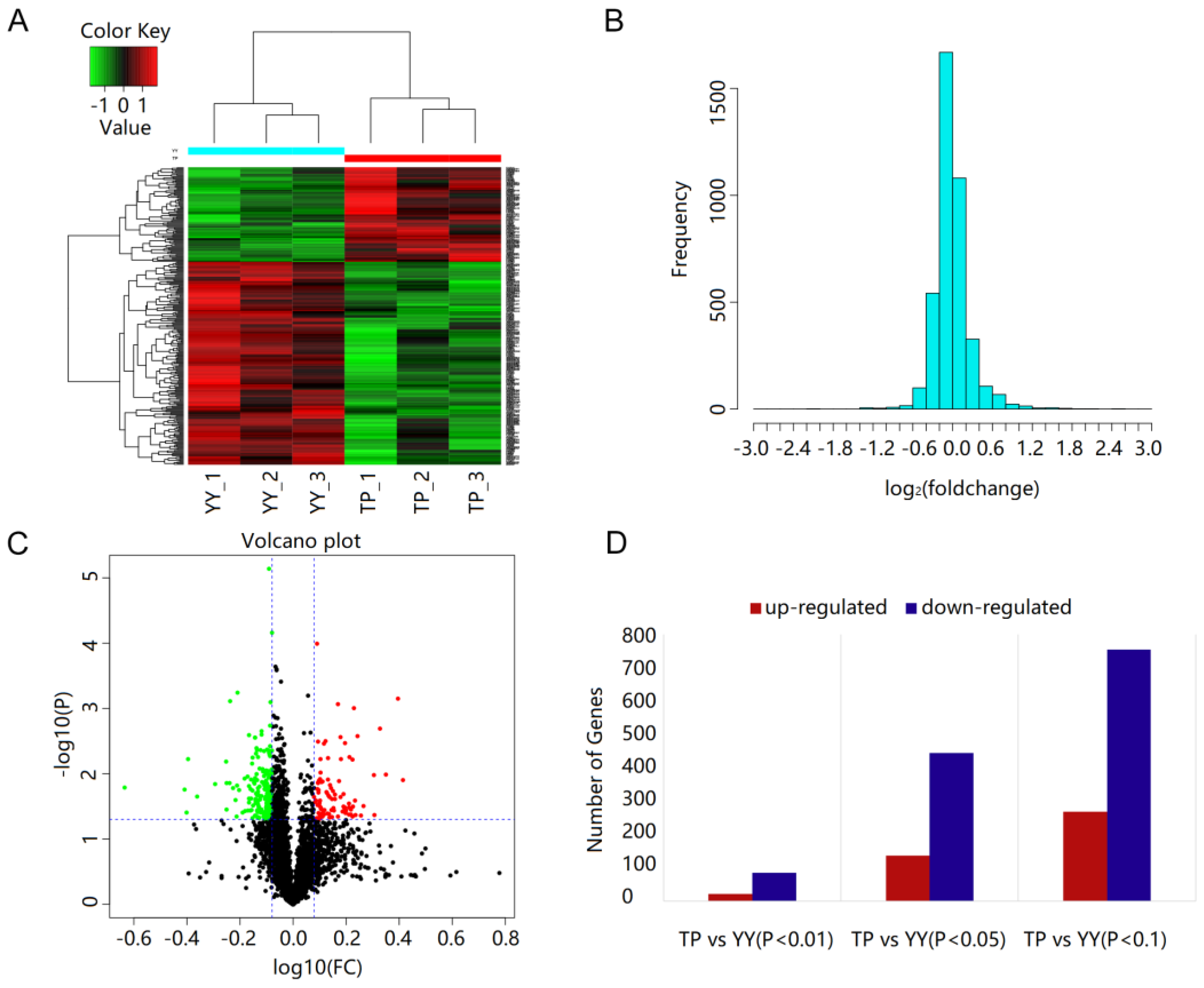
3.4. Protein Identification and Quantification
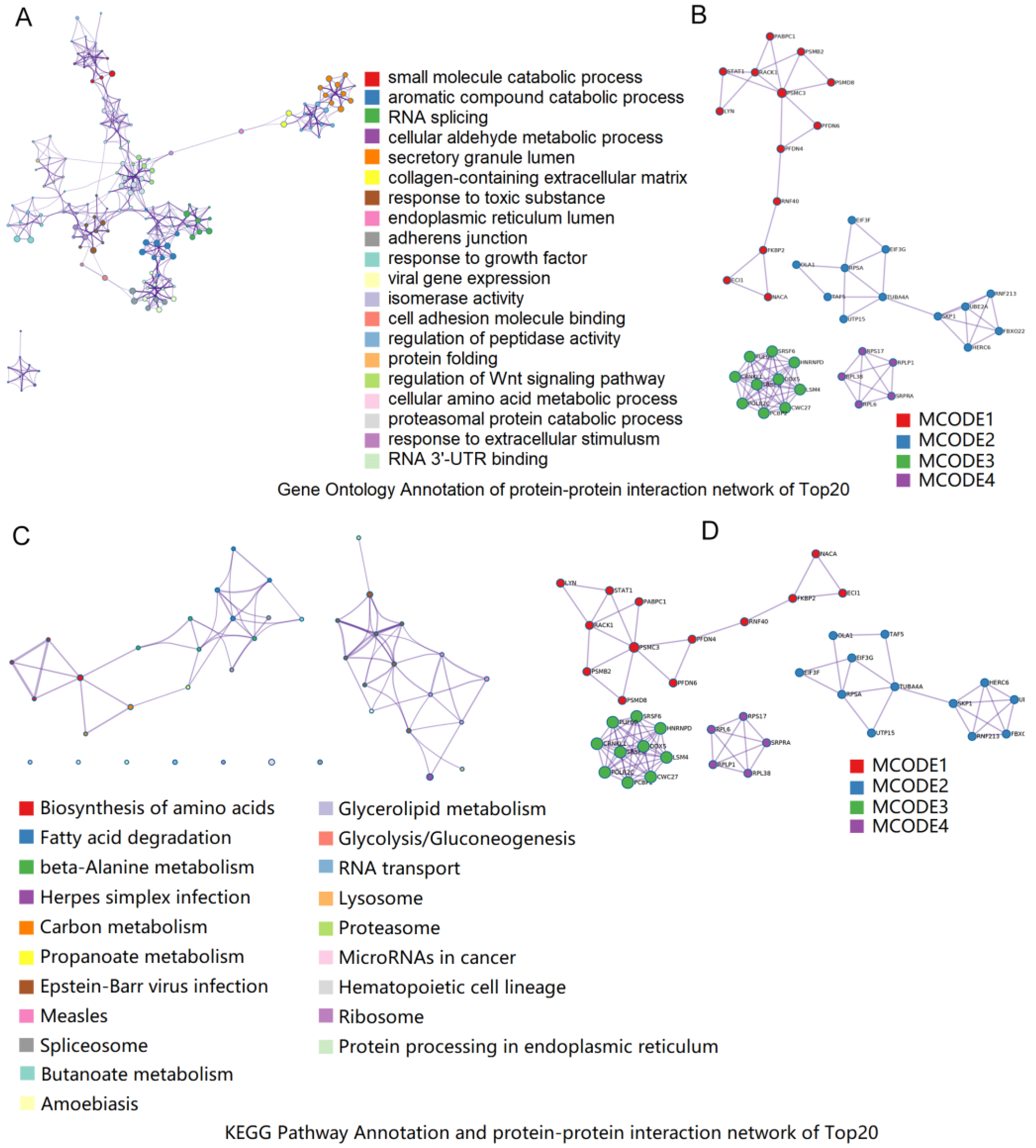
3.5. Functional Annotation of DEPs
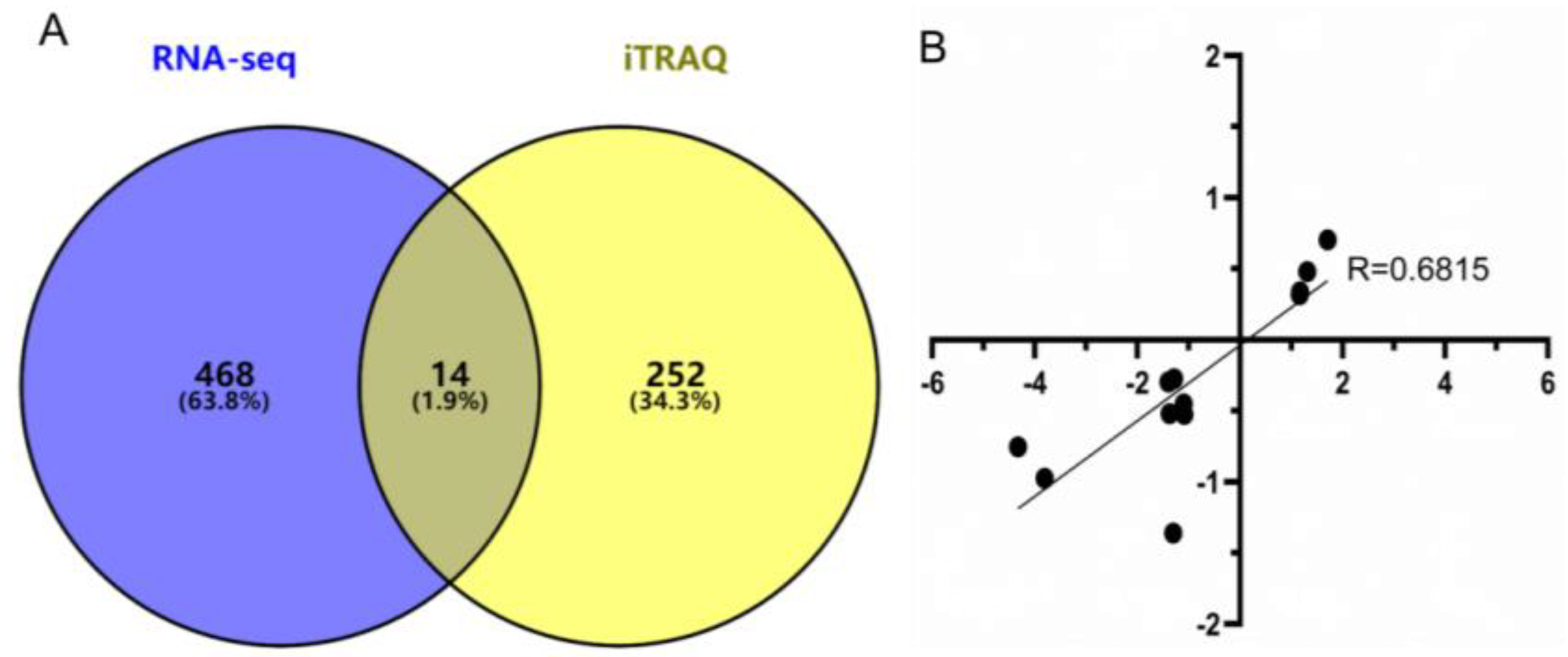
3.6. Combined Analysis of DEGs in RNA-seq and DEPs in iTRAQ
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fratantonio, D.; Cimino, F.; Speciale, A.; Virgili, F. Need (more than) two to tango, multiple tools to adapt to changes in oxygen availability. Biofactors 2018, 44, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Carey, D.; Thanaj, M.; Davies, T.; Gilber-Kawai, E.; Mitchell, K.; Levett, D.Z.H.; Mythen, M.G.; Martin, D.S.; Grocott, M.P.; Chipperfield, A.J.; et al. Enhanced flow-motion complexity of skin microvascular perfusion in Sherpas and lowlanders during ascent to high altitude. Sci. Rep. 2019, 9, 14391. [Google Scholar] [CrossRef] [Green Version]
- Lan, D.L.; Xiong, X.R.; Ji, W.H.; Li, J.; Mipam, T.D.; Ai, Y.; Chai, Z.X. Transcriptome profile and unique genetic evolution of positively selected genes in yak lungs. Genetica. 2018, 146, 151–160. [Google Scholar] [CrossRef]
- Guo, C.; Xu, J.; Wang, M.; Yan, T.; Yang, L.; Sun, Z. Study on oxygen supply standard for physical health of construction personnel of high-altitude tunnels. Int. J. Environ. Res. Public Health 2015, 13, 64. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.I.Y.; Ibrahim, M.E.; Khalil, E.A.G. High altitude and pre-eclampsia, adaptation or protection. Med. Hypotheses 2017, 104, 128–132. [Google Scholar] [CrossRef]
- Julian, C.G. Epigenomics and human adaptation to high altitude. J. Appl. Physiol. 2017, 123, 1362–1370. [Google Scholar] [CrossRef]
- Wu, T. Life on the high Tibetan Plateau. High. Alt. Med. Biol. 2004, 5, 1–2. [Google Scholar] [CrossRef]
- Kong, X.; Dong, X.; Yang, S.; Qian, J.; Yang, J.; Jiang, Q.; Li, X.; Wang, B.; Yan, D.; Lu, S.; et al. Natural selection on TMPRSS6 associated with the blunted erythropoiesis and improved blood viscosity in Tibetan pigs. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2019, 233, 11–22. [Google Scholar] [CrossRef]
- Jin, L.; Mao, K.; Li, J.; Huang, W.; Che, T.; Yuhua, F.; Tang, Q.; Liu, P.; Song, Y.; Liu, R.; et al. Genome-wide profiling of gene expression and DNA methylation provides insight into low-altitude acclimation in Tibetan pigs. Gene 2018, 642, 522–532. [Google Scholar] [CrossRef]
- Jia, C.; Kong, X.; Koltes, J.E.; Gou, X.; Yang, S.; Yan, D.; Lu, S. Gene co-expression network analysis unraveling transcriptional regulation of high-altitude adaptation of Tibetan Pig. PLoS ONE 2016, 11, e0168161. [Google Scholar] [CrossRef]
- Sharma, P.; Bansal, A.; Sharma, P.C. RNA-seq-based transcriptome profiling reveals differential gene expression in the lungs of Sprague-Dawley rats during early-phase acute hypobaric hypoxia. Mol. Genet. Genomics 2015, 290, 2225–2240. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Qin, C.; Xin, Y.; Yang, Y.; Hongguang, C.; Guolin, W.; Keliang, X.; Yonghao, Y. Itraq-based quantitative proteomic analysis of lungs in murine polymicrobial sepsis with hydrogen gas treatment. Shock 2018, 49, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chamba, Y.S.P.; Wang, Z.; Ma, J.; Wang, L.; Zhang, H. Comparative transcriptomic and proteomic analyses provide insights into the key genes involved in high-altitude adaptation in the Tibetan pig. Sci. Rep. 2017, 7, 3654. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Herrera-Marcos, L.V.; Lou-Bonafonte, J.M.; Martinez-Gracia, M.V.; Arnal, C.; Navarro, M.A.; Osada, J. Prenylcysteine oxidase 1, a pro-oxidant enzyme of low density lipoproteins. Front. Biosci. 2018, 23, 1020–1037. [Google Scholar]
- Han, L.; Huang, C.; Zhang, S. The RNA-binding protein SORBS2 suppresses hepatocellular carcinoma tumourigenesis and metastasis by stabilizing RORA mRNA. Liver Int. 2019, 39, 2190–2203. [Google Scholar] [CrossRef]
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- Silberman, A.; Goldman, O.; Boukobza Assayag, O.; Jacob, A.; Rabinovich, S.; Adler, L.; Lee, J.S.; Keshet, R.; Sarver, A.; Frug, J. Acid-Induced downregulation of ASS1 Contributes to the Maintenance of Intracellular pH in Cancer. Cancer Res. 2019, 79, 518–533. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.B.; Long, Y.; Chang, J.T.; Savaraj, N.; Feun, L.G.; Jung, M.; Chen, H.W.W.H. Chromatin remodeling system p300-HDAC2-Sin3A is involved in arginine starvation-induced hif-1α degradation at the ASS1 promoter for ASS1 Derepression. Sci. Rep. 2017, 7, 10814. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, Z.; Wang, Y.; Wei, L.; Chen, H. Overexpressed long noncoding RNA CPS1-IT alleviates pulmonary arterial hypertension in obstructive sleep apnea by reducing interleukin-1β expression via HIF1 transcriptional activity. J. Cell. Physiol. 2019, 234, 19715–19727. [Google Scholar] [CrossRef]
- Zhang, R.; Zhou, L.; Li, Q.; Liu, Q.; Yao, W.; Wan, H. Up-regulation of two actin-associated proteins prompts pulmonary artery smooth muscle cell migration under hypoxia. Am. J. Respir. Cell Mol. Biol. 2009, 41, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Kühn-Heid, E.C.D.; Kühn, E.C.; Ney, J.; Wendt, S.; Seelig, J.; Schwiebert, C.; Minich, W.D.; Stoppe, J.; Schomburg, L. Selenium-binding protein 1 indicates myocardial stress and risk for adverse outcome in cardiac surgery. Nutrients 2019, 11, 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonson, T.S.; McClain, D.A.; Jorde, L.B.; Prchal, J.T. Genetic determinants of Tibetan high-altitude adaptation. Hum. Genet. 2012, 131, 527–533. [Google Scholar] [CrossRef]
- Yang, Y.; Yuan, H.; Yang, q.; Cai, Y.; Ren, Y.; Li, Y.; Gao, C.; Zhao, S. Post-transcriptional regulation through alternative splicing in the lungs of Tibetan pigs under hypoxia. Gene 2022, 819, 146268. [Google Scholar] [CrossRef] [PubMed]
- Vohwinkel, C.U.; Hoegl, S.; Eltzschig, H.K. Hypoxia signaling during acute lung injury. J. Appl. Physiol. 2015, 119, 1157–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Gao, C.; Yang, T.; Sha, Y.; Cai, Y.; Wang, X.; Yang, Q.; Liu, C.; Wang, B.; Zhao, S. Vascular characteristics and expression of hypoxia genes in Tibetan pigs’ hearts. Vet. Med. Sci. 2021, 8, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Tian, S.; Jin, L.; Zhou, G.; Li, Y.; Zhang, Y.; Wang, T.; Yeung, C.K.L.; Chen, L.; Ma, J.; et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Gen. 2013, 45, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, Y.; Liu, S.; Zhang, C.; Cui, T.; Ding, K.; Wang, P.; Wang, X.; Wang, Z. KLF4, a key regulator of a transitive triplet, acts on the tgf-β signaling pathway and contributes to high-altitude adaptation of Tibetan Pigs. Front. Genet. 2021, 12, 6281932. [Google Scholar] [CrossRef]
- Ma, Y.F.; Han, X.M.; Huang, C.P.; Zhong, L.; Adeola, A.C.; Irwin, D.M.; Xie, H.B.; Zhang, Y.P. Population genomics analysis revealed origin and high-altitude adaptation of Tibetan pigs. Sci. Rep. 2019, 9, 11463. [Google Scholar] [CrossRef]
- Dong, K.; Yao, N.; Pu, Y.; He, X.; Zhao, Q.; Luan, Y.; Guan, W. Genomic scan reveals loci under altitude adaptation in Tibetan and Dahe pigs. PLoS ONE 2014, 9, e110520. [Google Scholar] [CrossRef]
- Mijalski, T.; Harder, A.; Halder, T.; Beckers, J. Identification of coexpressed gene clusters in a comparative analysis of transcriptome and proteome in mouse tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 8621–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Huang, J.; Zhong, Q.; She, Y.; Ou, R.; Li, C.; Chen, R.; Yao, M.; Zhang, Q.; Liu, S. Network-based analysis of the molecular mechanisms of multiple myeloma and monoclonal gammopathy of undetermined significance. Oncol. Lett. 2017, 14, 4167–4175. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhang, R.; Ye, S.; Liu, F.; Jiang, P.; Yu, X.; Xu, J.; Ma, L.; Cao, H.; Shen, Y.; et al. Alterations of human plasma proteome profile on adaptation to high-altitude hypobaric hypoxia. J. Proteome. Res. 2019, 18, 2021–2031. [Google Scholar] [CrossRef]
- Priyanka, A.; Sindhu, G.; Shyni, G.L.; Preetha Rani, M.R.; Nisha, V.M.; Raghu, K.G. Bilobalide abates inflammation, insulin resistance and secretion of angiogenic factors induced by hypoxia in 3T3-L1 adipocytes by controlling NF-κB and JNK activation. Int. Immunopharmacol. 2017, 42, 209–217. [Google Scholar] [CrossRef]
- Priyanka, A.; Nisha, V.M.; Anusree, S.S.; Raghu, K.G. Bilobalide attenuates hypoxia induced oxidative stress, inflammation, and mitochondrial dysfunctions in 3T3-L1 adipocytes via its antioxidant potential. Free Radic. Res. 2014, 48, 1206–1217. [Google Scholar] [CrossRef]
- Taglieri, L.; Nardo, T.; Vicinanza, R.; Ross, J.M.; Scarpa, S.; Coppotelli, G. Thyroid hormone regulates fibronectin expression through the activation of the hypoxia inducible factor 1. Biochem. Biophys. Res. Commun. 2017, 493, 1304–1310. [Google Scholar] [CrossRef]
- Simonides, W.S.; Mulcahey, M.A.; Redout, E.M.; Muller, A.; Zuidwijk, M.J.; Visser, T.J.; Wassen, F.W.J.S.; Crescenzi, A.; da-Silva, W.S.; Harney, J.; et al. Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J. Clin. Invest. 2008, 118, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Wu, Y.X.; Qiu, Y.B.; Wan, B.B.; Liu, G.; Chen, J.L.; Lu, M.D.; Pang, Q.F. Hyperoside suppresses hypoxia-induced A549 survival and proliferation through ferrous accumulation via AMPK/HO-1 axis. Phytomedicine 2019, 67, 153138. [Google Scholar] [CrossRef]
- Hong, C.F.; Chen, W.Y.; Wu, C.W. Upregulation of Wnt signaling under hypoxia promotes lung cancer progression. Oncol. Rep. 2017, 38, 1706–1714. [Google Scholar] [CrossRef] [Green Version]
- Scholten, D.J.; Timmer, C.M.; Peacock, J.D.; Pelle, D.W.; Williams, B.O.; Steensma, M.R. Down regulation of Wnt signaling mitigates hypoxia-induced chemoresistance in human osteosarcoma cells. PLoS ONE 2014, 9, e111431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Zhang, Y.; Zhang, S.; Wang, F.; Zhang, K.; Huang, Y.; Hour, Z.; Huang, G.; Wang, J. Oxymatrine enhanced anti-tumor effects of Bevacizumab against triple-negative breast cancer via abating Wnt/β-Catenin signaling pathway. Am. J. Cancer Res. 2019, 9, 1796–1814. [Google Scholar] [PubMed]
- Machuka, E.M.; Muigai, A.W.T.; Amimo, J.O.; Domelevo Entfellner, J.B.; Lekolool, I.; Abworo, E.O.; Pelle, R. Comparative analysis of SLA-1, SLA-2, and DQB1 genetic diversity in locally-adapted Kenyan pigs and their wild relatives, warthogs. Vet. Sci. 2021, 8, 180. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Martin, R.J.; Kladney, R.D.; Neumann, R.G.; Arbett, J.M. Hypoxia-inducible factor-1alpha suppresses squamous carcinogenic progression and epithelial-mesenchymal transition. Cancer Res. 2009, 69, 2638–2646. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.Y.; Zhou, J.R.; Gao, C.; Feldman, L.; Sytkowski, A.J. Human selenium binding protein-1 (hSP56) is a negative regulator of HIF-1α and suppresses the malignant characteristics of prostate cancer cells. BMB Rep. 2014, 47, 411–416. [Google Scholar] [CrossRef] [Green Version]
- Young, T.; Poobalan, Y.; Tan, E.K.; Tao, S.; Ong, S.; Wehner, P.; Schwenty-Lara, J.; Lim, C.Y.; Sadasivam, A.; Lovatt, M. The PDZ domain protein Mcc is a novel effector of non-canonical Wnt signaling during convergence and extension in zebrafish. Development 2014, 141, 3505–3516. [Google Scholar] [CrossRef] [Green Version]
- Dunn, N.R.; Tolwinski, N.S. Ptk7 and Mcc, unfancied components in non-canonical Wnt signaling and cancer. Cancers 2016, 8, 68. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Mitomi, H.; Saito, T.; Takahashi, M.; Sakamoto, N.; Fukui, N.; Yao, T.; Watanabe, S. Distinct WNT/β-catenin signaling activation in the serrated neoplasia pathway and the adenoma-carcinoma sequence of the colorectum. Mod. Pathol. 2015, 28, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Hu, H.; Wang, X.; Ye, W.; Su, H.; Hu, Y.; Dong, L.; Zhang, R.; Ying, K. Involvement of CapG in proliferation and apoptosis of pulmonary arterial smooth muscle cells and in hypoxia-induced pulmonary hypertension rat model. Exp. Lung Res. 2016, 42, 142–153. [Google Scholar] [CrossRef]
- Liao, S.H.; Zhao, X.Y.; Han, Y.H.; Zhang, J.; Wang, L.S.; Xia, L.; Zhao, K.W.; Zheng, Y.; Guo, M.; Chen, G.Q. Proteomics-based identification of two novel direct targets of hypoxia-inducible factor-1 and their potential roles in migration/invasion of cancer cells. Proteomics 2009, 9, 3901–3912. [Google Scholar] [CrossRef]
- Long, Y.; Tsai, W.B.; Chang, J.T.; Estecio, M.; Wangpaichitr, M.; Savaraj, N.; Feun, L.G.; Chen, H.H.W.; Kuo, M.T. Cisplatin-induced synthetic lethality to arginine-starvation therapy by transcriptional suppression of ASS1 is regulated by DEC1, HIF-1α, and c-Myc transcription network and is independent of ASS1 promoter DNA methylation. Oncotarget 2016, 7, 82658–82670. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.R.; Zhang, M.Y.; Rao, H.L.; Pu, H.Y.; Zhang, H.Z.; Wang, H.Y. Identification of ADH4 as a novel and potential prognostic marker in hepatocellular carcinoma. Med. Oncol. 2012, 29, 2737–2743. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Tabei, H.; Iwahuti, M. Characterization of low-acetic-acid-producing yeast isolated from 2-deoxyglucose-resistant mutants and its application to high-gravity brewing. J. Biosci. Bioeng. 2006, 101, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wu, M.; Tang, G.P.; Wang, H.J.; Huang, C.X.; Wu, X.J.; He, Y.; Zhang, B.; Huang, C.H.; Liu, H.; et al. Megalobrama amblycephala effects of acute hypoxia and reoxygenation on physiological and immune responses and redox balance of wuchang bream (Yih, 1955). Front. Physiol. 2017, 8, 375. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Yuan, W.; Song, J.; Wang, S.; Gu, X. LncRNA CPS1-IT1 suppresses EMT and metastasis of colorectal cancer by inhibiting hypoxia-induced autophagy through inactivation of HIF-1α. Biochimie 2018, 144, 21–27. [Google Scholar] [CrossRef]
- Çeliktas, M.; Tanaka, I.; Tripathi, S.C.; Fahrmann, J.F.; Aguilar-Bonavides, C.; Villalobos, P.; Delgado, O.; Dhillon, D.; Dennison, J.B.; Ostrin, E.J.; et al. Role of CPS1 in cell growth, metabolism and prognosis inLKB1-Inactivated Lung Adenocarcinoma. J. Natl. Cancer Inst. 2017, 109, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciumas, M.; Eyries, M.; Poirier, O.; Maugenre, S.; Dierick, F.; Gambaryan, N.; Montagne, K.; Nadaud, S.; Soubrier, F. Bone morphogenetic proteins protect pulmonary microvascular endothelial cells from apoptosis by upregulating α-B-crystallin. Arterioscler Thromb. Vasc. Biol. 2013, 33, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Snyder, B.; Wu, H.K.; Tillman, B.; Floyd, T.F. Aged Mouse Hippocampus Exhibits Signs of Chronic Hypoxia and an Impaired HIF-Controlled Response to Acute Hypoxic Exposures. Cells 2022, 11, 423. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Name | Log2 Fold Change | p-Value | Up/Down |
|---|---|---|---|---|
| ENSSSCG00000009738 | GALNT9 | 7.689227 | 1.83 × 10−6 | up |
| ENSSSCG00000033254 | IFN-ALPHA-13 | 6.983059 | 1.02 × 10−6 | |
| ENSSSCG00000007859 | UMOD | 6.040727 | 0.009784 | |
| ENSSSCG00000033610 | ZG16B | 6.015486 | 0.024330 | |
| ENSSSCG00000037533 | HIST2H2AB | 5.656798 | 0.001126 | |
| ENSSSCG00000029516 | SLC22A8 | 5.639100 | 0.020157 | |
| ENSSSCG00000034995 | RF00017 | 5.098178 | 0.035442 | |
| ENSSSCG00000037202 | CACNG4 | 4.860514 | 0.013594 | |
| ENSSSCG00000037225 | RPRM | 4.856914 | 0.016825 | |
| ENSSSCG00000008741 | FGFBP1 | 4.832671 | 0.000081 | |
| ENSSSCG00000037535 | SLC34A1 | 4.729766 | 0.013466 | |
| ENSSSCG00000037300 | GRP | 4.443211 | 0.005612 | |
| ENSSSCG00000035689 | NEXMIF | 4.430799 | 0.043955 | |
| ENSSSCG00000001906 | CYP1A1 | 4.426375 | 0.000505 | |
| ENSSSCG00000003066 | IRGC | 4.404553 | 0.047451 | |
| ENSSSCG00000033193 | TPO | 4.400171 | 0.016735 | |
| ENSSSCG00000037534 | OPCML | 4.233214 | 4.60 × 10−16 | |
| ENSSSCG00000001613 | TREML1 | 4.174212 | 0.003859 | |
| ENSSSCG00000028695 | TMSB15A | 4.032314 | 3.94 × 10−14 | |
| ENSSSCG00000022140 | TMPRSS11E | 4.005328 | 0.028356 | |
| ENSSSCG00000040910 | APOH | −4.214353 | 0.027279 | down |
| ENSSSCG00000021767 | TDH | −4.268103 | 0.001937 | |
| ENSSSCG00000002479 | SERPINA11 | −4.286988 | 0.006838 | |
| ENSSSCG00000012711 | F9 | −4.323437 | 0.026465 | |
| ENSSSCG00000016159 | CPS1 | −4.326881 | 0.000381 | |
| ENSSSCG00000012517 | TMSB15B | −4.341481 | 5.88 × 10−7 | |
| ENSSSCG00000016856 | C9 | −4.466406 | 0.037223 | |
| ENSSSCG00000010431 | A1CF | −4.487968 | 0.017588 | |
| ENSSSCG00000003835 | C8A | −4.594111 | 0.000224 | |
| ENSSSCG00000020680 | CLDN14 | −4.631278 | 0.006969 | |
| ENSSSCG00000008998 | FGA | −4.729940 | 0.002310 | |
| ENSSSCG00000036158 | TRAM1L1 | −4.734145 | 0.003310 | |
| ENSSSCG00000037547 | SLC17A3 | −4.893193 | 0.000000 | |
| ENSSSCG00000006248 | MOS | −5.303923 | 0.006916 | |
| ENSSSCG00000002983 | LGALS13 | −5.369302 | 0.000084 | |
| ENSSSCG00000034429 | PLA2G5 | −5.410249 | 0.010311 | |
| ENSSSCG00000029449 | PRG4 | −5.667579 | 4.54 × 10−20 | |
| ENSSSCG00000016315 | SPP2 | −6.047557 | 0.000038 | |
| ENSSSCG00000008214 | FABP1 | −6.452786 | 0.000014 | |
| ENSSSCG00000037268 | APCS | −7.280566 | 2.05 × 10−7 |
| Accession | Gene Name | MW [kDa] | FC | p-Value | Log2FC | |
|---|---|---|---|---|---|---|
| A0A286ZWS8 | COL2A1 | 141.6 | 4.119454 | 0.321115 | 2.042453 | up |
| F8WSC1 | SLA-1 | 40 | 3.918033 | 0.357562 | 1.970129 | |
| V9PR54 | SLA-1 | 40.4 | 3.158697 | 0.138104 | 1.659330 | |
| A0A287AEL2 | KRT14 | 56 | 3.135079 | 0.288655 | 1.648502 | |
| A0A287ATD0 | KRT75 | 58.7 | 3.059540 | 0.166403 | 1.613315 | |
| I3L8B2 | COL9A2 | 65.1 | 2.900520 | 0.371928 | 1.536312 | |
| A0A287B863 | ACAN | 251.9 | 2.875969 | 0.352952 | 1.524048 | |
| F1S571 | COL11A1 | 147.1 | 2.861647 | 0.081541 | 1.516846 | |
| F1REZ1 | HAPLN1 | 40.2 | 2.843049 | 0.359189 | 1.507439 | |
| A0A286ZI25 | PARP14 | 200.9 | 2.780718 | 0.354096 | 1.475458 | |
| F2Z501 | TMED2 | 21.7 | 2.649027 | 0.073811 | 1.405462 | |
| F1S0J1 | C4BPA | 22.7 | 2.597122 | 0.012400 | 1.376914 | |
| F1SCU3 | MATN3 | 52.7 | 2.556017 | 0.376462 | 1.353897 | |
| F1RXG1 | KRT27 | 49.7 | 2.384095 | 0.227473 | 1.253442 | |
| A5A758 | KRT1 | 65.2 | 2.333889 | 0.126611 | 1.222736 | |
| A0A287A461 | CHAD | 40.6 | 2.294893 | 0.355746 | 1.198427 | |
| F1S7K4 | PLIN4 | 158.7 | 2.237992 | 0.010325 | 1.162205 | |
| I3LDS3 | KRT10 | 58.9 | 2.234501 | 0.340817 | 1.159953 | |
| I3L5Q7 | MATN1 | 53.9 | 2.227542 | 0.361420 | 1.155452 | |
| A7J149 | BPIFB1 | 51.9 | 2.227004 | 0.377700 | 1.155104 | |
| F1SIT7 | RPLP1 | 11.5 | 0.576307 | 0.013941 | −0.795091 | down |
| F1RW28 | HSD17B13 | 33.3 | 0.561833 | 0.013894 | −0.831787 | |
| F1RL41 | UPB1 | 42.9 | 0.560874 | 0.035026 | −0.834251 | |
| A0A287BN06 | PZP | 158.1 | 0.560354 | 0.006529 | −0.835590 | |
| B5L2L8 | SLA-DQA | 9.5 | 0.546931 | 0.058481 | −0.870568 | |
| A0A0A7BZH1 | SLA-DQB1 | 29.5 | 0.538462 | 0.365487 | −0.893085 | |
| L7UWL8 | SLA-2 | 20.7 | 0.538067 | 0.396005 | −0.894142 | |
| A0A2C9F382 | FABP1 | 16.5 | 0.537673 | 0.051562 | −0.895199 | |
| A0A2C9F343 | LYZ | 16.5 | 0.508926 | 0.014242 | −0.974471 | |
| Q8HX61 | SLA-B | 40.6 | 0.482827 | 0.230141 | −1.050422 | |
| B6DU23 | SLA-DRB1 | 10.8 | 0.470833 | 0.319788 | −1.086712 | |
| B6ICW6 | SLA-2 | 40.5 | 0.445435 | 0.385493 | −1.166714 | |
| D3GIN8 | SLA-2 | 38.8 | 0.435303 | 0.022197 | −1.199910 | |
| K9J6H8 | A2M | 163.9 | 0.430956 | 0.069842 | −1.214386 | |
| A0A1L1YNR3 | FASN | 93.4 | 0.423150 | 0.060034 | −1.240759 | |
| T2HGI4 | SLA-1 | 35 | 0.404494 | 0.333977 | −1.305808 | |
| D3GIP1 | SLA-3 | 40.4 | 0.402103 | 0.005976 | −1.314364 | |
| I3LLB7 | PIK3R1 | 83.5 | 0.396648 | 0.039119 | −1.334069 | |
| F1SER3 | SFTPA1 | 26.5 | 0.389442 | 0.017424 | −1.360520 | |
| A0A1C9J6L2 | SLA-1 | 40.1 | 0.231828 | 0.016153 | −2.108876 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, P.; Zhang, B.; Li, P.; Ahmed, Z.; Hu, X.; Chamba, Y.; Zhang, H. Plateau Adaptation Gene Analyses Reveal Transcriptomic, Proteomic, and Dual Omics Expression in the Lung Tissues of Tibetan and Yorkshire Pigs. Animals 2022, 12, 1919. https://doi.org/10.3390/ani12151919
Shang P, Zhang B, Li P, Ahmed Z, Hu X, Chamba Y, Zhang H. Plateau Adaptation Gene Analyses Reveal Transcriptomic, Proteomic, and Dual Omics Expression in the Lung Tissues of Tibetan and Yorkshire Pigs. Animals. 2022; 12(15):1919. https://doi.org/10.3390/ani12151919
Chicago/Turabian StyleShang, Peng, Bo Zhang, Pan Li, Zulfiqar Ahmed, Xiaoxiang Hu, Yangzom Chamba, and Hao Zhang. 2022. "Plateau Adaptation Gene Analyses Reveal Transcriptomic, Proteomic, and Dual Omics Expression in the Lung Tissues of Tibetan and Yorkshire Pigs" Animals 12, no. 15: 1919. https://doi.org/10.3390/ani12151919