
De Novo Whole-Genome Sequencing and Assembly of the Yellow-Throated Bunting (Emberiza elegans) Provides Insights into Its Evolutionary Adaptation

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
2.2. Genome Survey and Assembly
2.3. Genome Annotation
2.4. Gene Family and Phylogenetic Analysis
2.5. Analysis of Positive Selection
2.6. Population Dynamics
3. Results
3.1. De Novo Genome-Sequencing Assembly and Assessment
3.2. Genome Annotation
3.3. Phylogenetic Relationships and Divergence Times
3.4. Gene Family Expansion and Positive Selection
3.5. Temporal Population Dynamics
4. Discussion
4.1. Genomic Features
4.2. Functional Assignment
4.3. Demographic History
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- MacKinnon, J. Guide to the Birds of China; Oxford University Press: London, UK, 2022. [Google Scholar]
- Zheng, G.M. A Checklist on the Classification and Distribution of the Birds of China, 2nd ed.; Science Press: Beijing, China, 2002. [Google Scholar]
- Nam, H.Y.; Choi, C.Y.; Park, J.G.; Hong, G.P.; Won, I.J.; Kim, S.J.; Bing, G.C.; Chae, H.Y. Protandrous migration and variation in morpho-logical characters in Emberiza buntings at an East Asian stopover site. Ibis 2011, 153, 494–501. [Google Scholar] [CrossRef]
- Cai, T.; Wu, G.; Sun, L.; Zhang, Y.; Peng, Z.; Guo, Y.; Liu, X.; Pan, T.; Chang, J.; Sun, Z.; et al. Biogeography and diversification of Old World buntings (Aves: Emberizidae): Radiation in open habitats. J. Avian Biol. 2021, 52, e02672. [Google Scholar] [CrossRef]
- Wang, S.R.; Joka, F.R.; Wang, X.L.; Bai, S.Y. Analysis of the Phylogeny and Evolutionary Selection Pressure of the Mx Gene in 10 Wild Birds. Pak. J. Zool. 2019, 51, 1299–1307. [Google Scholar] [CrossRef]
- Wang, Y.H.; Wu, W.; An, Y.X.; Zhang, W.; Xu, Q. Protandry in Yellow-Throated Bunting (Emberiza elegans) During Spring in the Maoershan Mountain region of Heilongjiang Province, China. Chin. J. Wildl. 2019, 40, 679–684. [Google Scholar]
- Bennetzen, J.L. Patterns in grass genome evolution. Curr. Opin. Plant Biol. 2007, 10, 176–181. [Google Scholar] [CrossRef]
- Stiller, J.; Zhang, G. Comparative Phylogenomics, a Stepping Stone for Bird Biodiversity Studies. Diversity 2019, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.H.; Shi, Y.J.; Yuan, J.Y.; Hu, X.S.; Zhang, H.; Li, N.; Li, Z.Y.; Chen, Y.X.; Mu, D.S.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2013, arXiv:1308.2012. [Google Scholar]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the Gap: Upgrading Genomes with Pacific Biosciences RS Long-Read Sequencing Technology. PLoS ONE 2012, 7, e47768. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Tarailo, G.M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioin-Form. 2009, 25, 4–10. [Google Scholar]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [Green Version]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA1. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [Green Version]
- Birney, E.; Durbin, R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 2000, 10, 547–548. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ri-bosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Griffiths, J.S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yu, L.; Kalavacharla, V.; Liu, Z. A Bayesian model for gene family evolution. BMC Bioinform. 2011, 12, 426. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RaxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L.; Edwards, S.V. A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol. Biol. 2010, 10, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ng, E.K.; Ng, Y.P.; Wong, C.Y.; Yu, J.; Jin, H.; Cheng, V.Y.; Go, M.Y.; Cheung, P.K.; Ebert, M.P.; et al. Identification of retinoic ac-id-regulated nuclear matrix-associated protein as a novel regulator of gastric cancer. Br. J. Cancer 2009, 101, 691. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo, G.A.; Schmitt, C.J.; Edwards, S.V. What have we learned from the first 500 avian genomes? Annu. Rev. Ecol. Evol. Syst. 2021, 52, 611–639. [Google Scholar] [CrossRef]
- Cai, Q.L.; Qian, X.J.; Lang, Y.S.; Luo, Y.D.; Xu, J.H.; Pan, S.K.; Hui, Y.Y.; Gou, C.Y.; Cai, Y.; Hao, M.R. Genome sequence of ground tit Pseu-dopodoces humilis and its adaptation to high altitude. Genome Biol. 2013, 14, R29. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Z.H.; Wang, X.Z.; Ding, M.X. Cell Biology, 4th ed.; High Education Press: Beijing, China, 2011. [Google Scholar]
- Niot, I.; Poirier, H.; Tran, T.T.T.; Besnard, P. Intestinal absorption of long-chain fatty acids: Evidence and uncertainties. Prog. Lipid Res. 2009, 48, 101–115. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Wong, M.M.; Fish, E.N. Chemokines: Attractive mediators of the immune response. Semin. Immunol. 2002, 15, 5–14. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Tkaczyk, C. Integrated signalling pathways for mast-cell activation. Nat. Rev. Immunol. 2006, 6, 218–230. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Gutkowska, J.; Jankowski, M.; Lambert, C.; Mukaddam-Daher, S.; Zingg, H.H.; McCann, S.M. Oxytocin releases atrial natriuretic peptide by combining with oxytocin receptors in the heart. Proc. Natl. Acad. Sci. USA 1997, 94, 11704–11709. [Google Scholar] [CrossRef] [Green Version]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef] [Green Version]
- Warren, W.C.; Clayton, D.F.; Ellegren, H.; Arnold, A.P.; Hillier, L.W.; Künstner, A.; Searle, S.; White, S.; Vilella, A.; Fairley, S.; et al. The genome of a songbird. Nature 2010, 464, 757–762. [Google Scholar] [CrossRef]
- Kapusta, A.; Suh, A. Evolution of bird genomes-a transposon’s-eye view. Ann. N. Y. Acad. Sci. 2016, 1389, 164–185. [Google Scholar] [CrossRef]
- Kapusta, A.; Suh, A.; Feschotte, C. The hidden elasticity of avian and mammalian genomes. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Boman, J.; Frankl-Vilches, C.; dos Santos, M.D.; de Oliveira, E.H.C.; Gahr, M.; Suh, A. The genome of Blue-Capped Cordon-Bleu Un-covers hidden diversity of LTR retrotransposons in Zebra Finch. Genes 2019, 10, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; The Avian Genome Consortium; Li, B.; Li, C.; Gilbert, M.T.P.; Jarvis, E.D.; Wang, J. Comparative genomic data of the Avian Phylogenomics Project. GigaScience 2014, 3, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmo, C.G.; Haunerland, N.H.; Hochachka, P.W.; Williams, T.D. Seasonal dynamics of flight muscle fatty acid binding protein and catabolic enzymes in a migratory shorebird. Am. J. Physiol. Integr. Comp. Physiol. 2002, 282, R1405–R1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corder, K.R.; DeMoranville, K.J.; Russell, D.E.; Huss, J.M.; Schaeffer, P.J. Annual life-stage regulation of lipid metabolism and storage and association with PPARs in the migrant species Gray Catbird (Dumetella carolinensis). J. Exp. Biol. 2016, 219, 3391–3398. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, X.; Chen, B.; Guo, Y.; Tang, H.; Li, D.; Liu, D.; Wang, Y.; Li, G.; Kang, X.; et al. Association of a new 99-bp indel of the CEL gene promoter region with phenotypic traits in chickens. Sci. Rep. 2020, 10, 3215. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Jiao, X.; Zhang, D.; Cheng, Y.; Song, G.; Qu, Y.; Lei, F. Comparative Genomics and Evolution of Avian Specialized Traits. Curr. Genom. 2021, 22, 496–511. [Google Scholar] [CrossRef]
- Johnston, R.A.; Paxton, K.L.; Moore, F.R.; Wayne, R.K.; Smith, T.B. Seasonal gene expression in a migratory songbird. Mol. Ecol. 2016, 25, 5680–5691. [Google Scholar] [CrossRef]
- Kumar, V.; Wingfield, J.C.; Dawson, A.; Ramenofsky, M.; Rani, S.; Bartell, P. Biological Clocks and Regulation of Seasonal Reproduction and Migration in Birds. Physiol. Biochem. Zool. 2010, 83, 827–835. [Google Scholar] [CrossRef]
- Yumimoto, K.; Muneoka, T.; Tsuboi, T.; Nakayama, K.I. Substrate Binding Promotes Formation of the Skp1-Cul1-Fbxl3 (SCFFbxl3) Protein Complex. J. Biol. Chem. 2013, 288, 32766–32776. [Google Scholar] [CrossRef] [Green Version]
- Figuerola, J.; Green, A.J. Haematozoan Parasites and Migratory Behaviour in Waterfowl. Evol. Ecol. 2000, 14, 143–153. [Google Scholar] [CrossRef]
- Nadachowska-Brzyska, K.; Li, C.; Smeds, L.; Zhang, G.; Ellegren, H. Temporal Dynamics of Avian Populations during Pleistocene Revealed by Whole-Genome Sequences. Curr. Biol. 2015, 25, 1375–1380. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; Wang, H.; Orozcoterwengel, P.; Hu, C.-C.; Wu, G.-Y.; Qian, L.-F.; Sun, Z.-L.; Shi, W.-B.; Yan, P.; Wu, X.-B.; et al. Long-term sky islands generate highly divergent lineages of a narrowly distributed stream salamander (Pachyhynobius shangchengensis) in mid-latitude mountains of East Asia. BMC Evol. Biol. 2019, 19, 1. [Google Scholar] [CrossRef]
- McCormack, J.; Huang, H.; Knowle, L. Sky islands. In Encyclopedia of Islands; Gillespie, R.G., Clague, D.A., Eds.; University of California Press: Berkeley, CA, USA, 2009; pp. 841–843. [Google Scholar]
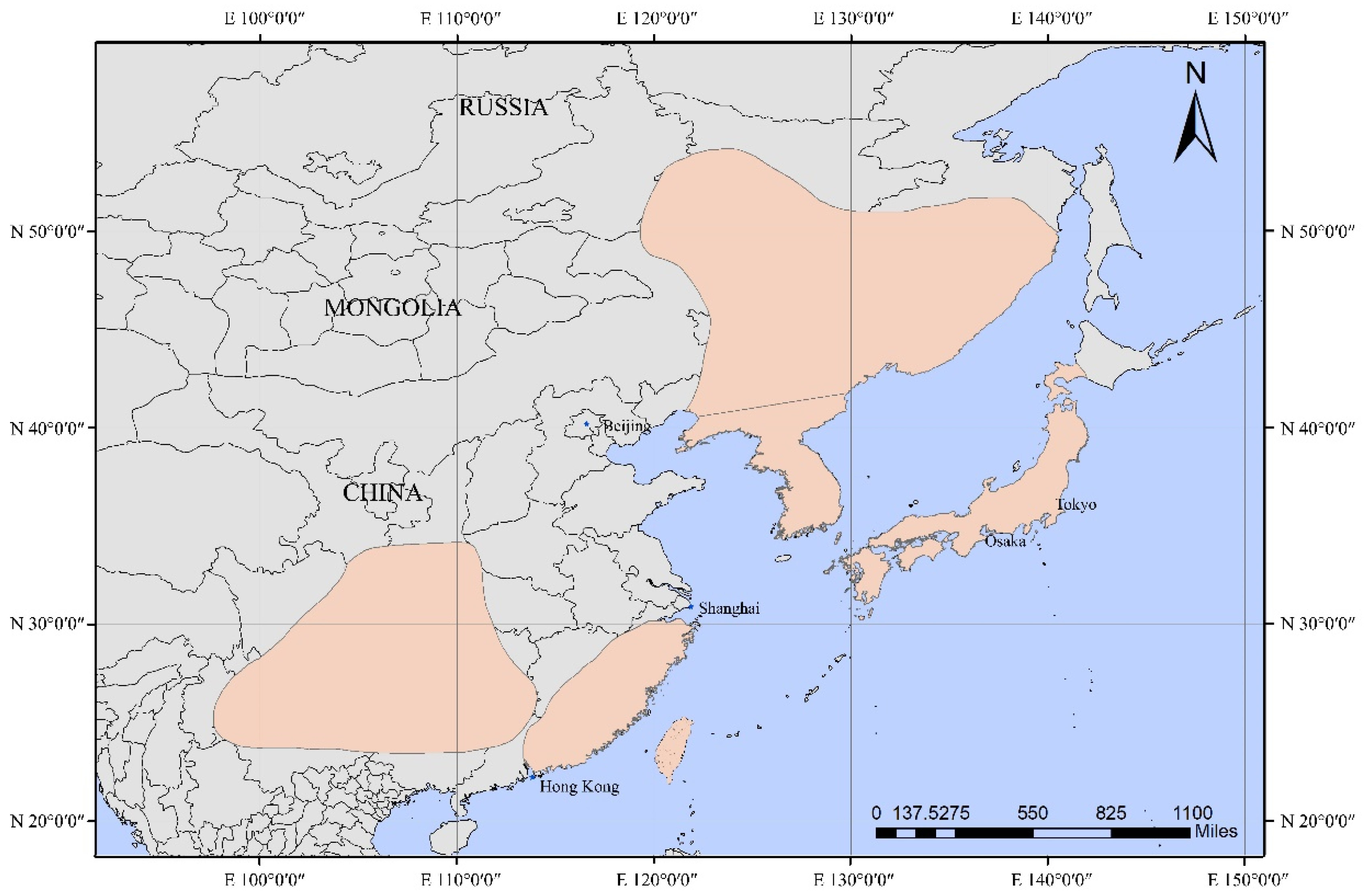
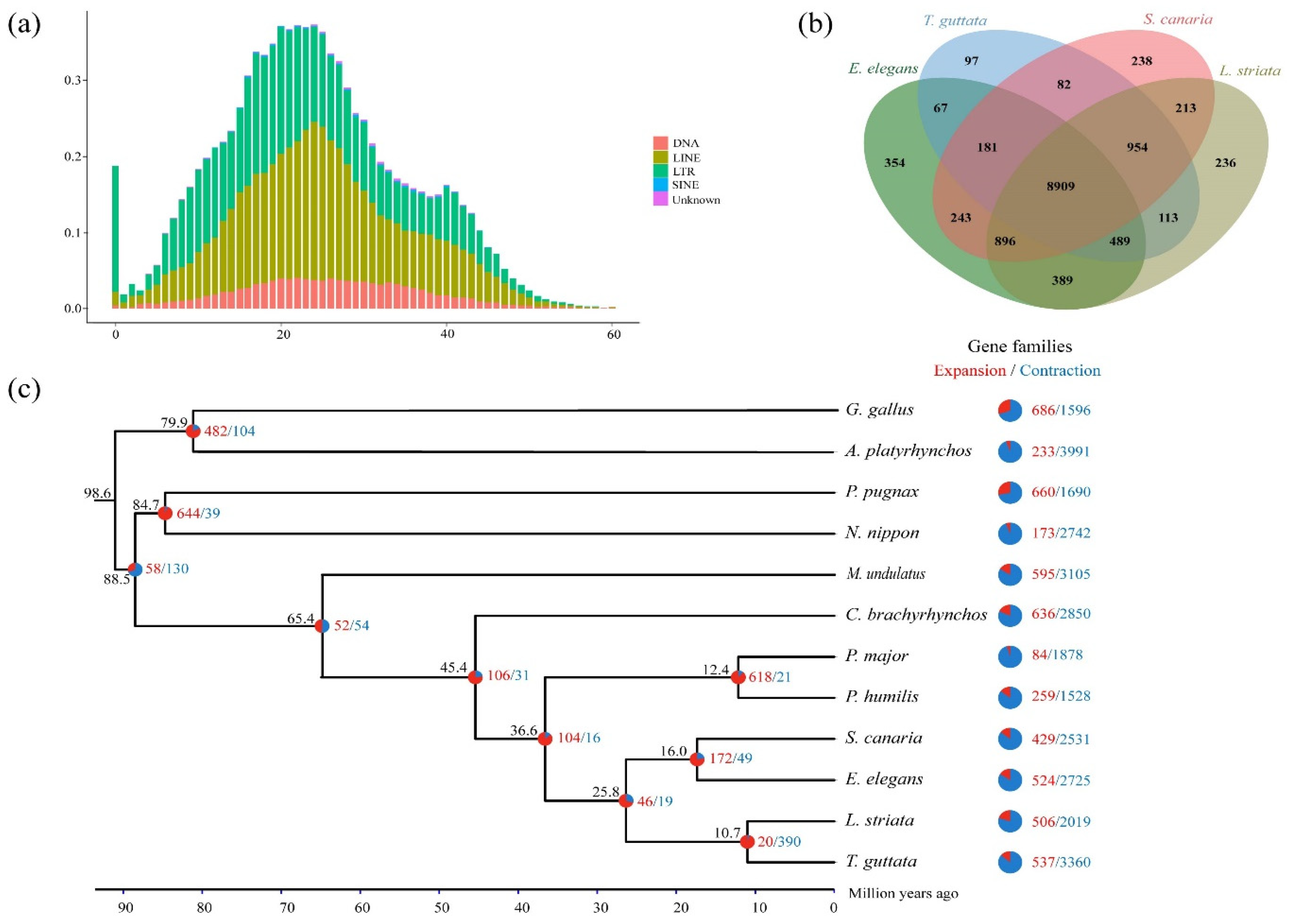


{kind=link}
{kind=link}
{kind=link}
| Contig | Scaffold | |
|---|---|---|
| Number | 4522 | 4026 |
| N50 | 16,287,717 | 28,938,815 |
| N90 | 1,224,325 | 2,340,365 |
| Total length | 1,141,058,764 | 1,144,605,635 |
| Species | Number | Average Transcript Length (bp) | Average CDS Length (bp) | Average Exons per Gene | Average Exon Length (bp) | Average Intron Length (bp) |
|---|---|---|---|---|---|---|
| Emberiza elegans | 15,868 | 25,516.83 | 1600.26 | 9.62 | 166.30 | 2773.68 |
| Gallus gallus | 17,444 | 29,696.28 | 1735.32 | 10.19 | 170.35 | 3043.54 |
| Lonchura striata | 15,420 | 28,472.72 | 1774.93 | 10.66 | 166.45 | 2762.73 |
| Parus major | 15,240 | 28,701.65 | 1802.84 | 10.76 | 167.49 | 2754.87 |
| Serinus canaria | 14,756 | 27,514.15 | 1729.92 | 10.74 | 161.08 | 2647.35 |
| Taeniopygia guttata | 16,351 | 26,215.51 | 1619.92 | 10.01 | 161.88 | 2730.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, T.; Chen, G.; Xu, Z.; Luo, S.; Wang, H.; Li, C.; Shan, L.; Zhang, B. De Novo Whole-Genome Sequencing and Assembly of the Yellow-Throated Bunting (Emberiza elegans) Provides Insights into Its Evolutionary Adaptation. Animals 2022, 12, 2004. https://doi.org/10.3390/ani12152004
Hu T, Chen G, Xu Z, Luo S, Wang H, Li C, Shan L, Zhang B. De Novo Whole-Genome Sequencing and Assembly of the Yellow-Throated Bunting (Emberiza elegans) Provides Insights into Its Evolutionary Adaptation. Animals. 2022; 12(15):2004. https://doi.org/10.3390/ani12152004
Chicago/Turabian StyleHu, Tingli, Guotao Chen, Zhen Xu, Site Luo, Hui Wang, Chunlin Li, Lei Shan, and Baowei Zhang. 2022. "De Novo Whole-Genome Sequencing and Assembly of the Yellow-Throated Bunting (Emberiza elegans) Provides Insights into Its Evolutionary Adaptation" Animals 12, no. 15: 2004. https://doi.org/10.3390/ani12152004
APA StyleHu, T., Chen, G., Xu, Z., Luo, S., Wang, H., Li, C., Shan, L., & Zhang, B. (2022). De Novo Whole-Genome Sequencing and Assembly of the Yellow-Throated Bunting (Emberiza elegans) Provides Insights into Its Evolutionary Adaptation. Animals, 12(15), 2004. https://doi.org/10.3390/ani12152004