
Identification and Characterization of Alternative Splicing Variants and Positive Selection Genes Related to Distinct Growth Rates of Antlers Using Comparative Transcriptome Sequencing
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Data and Sample Collection
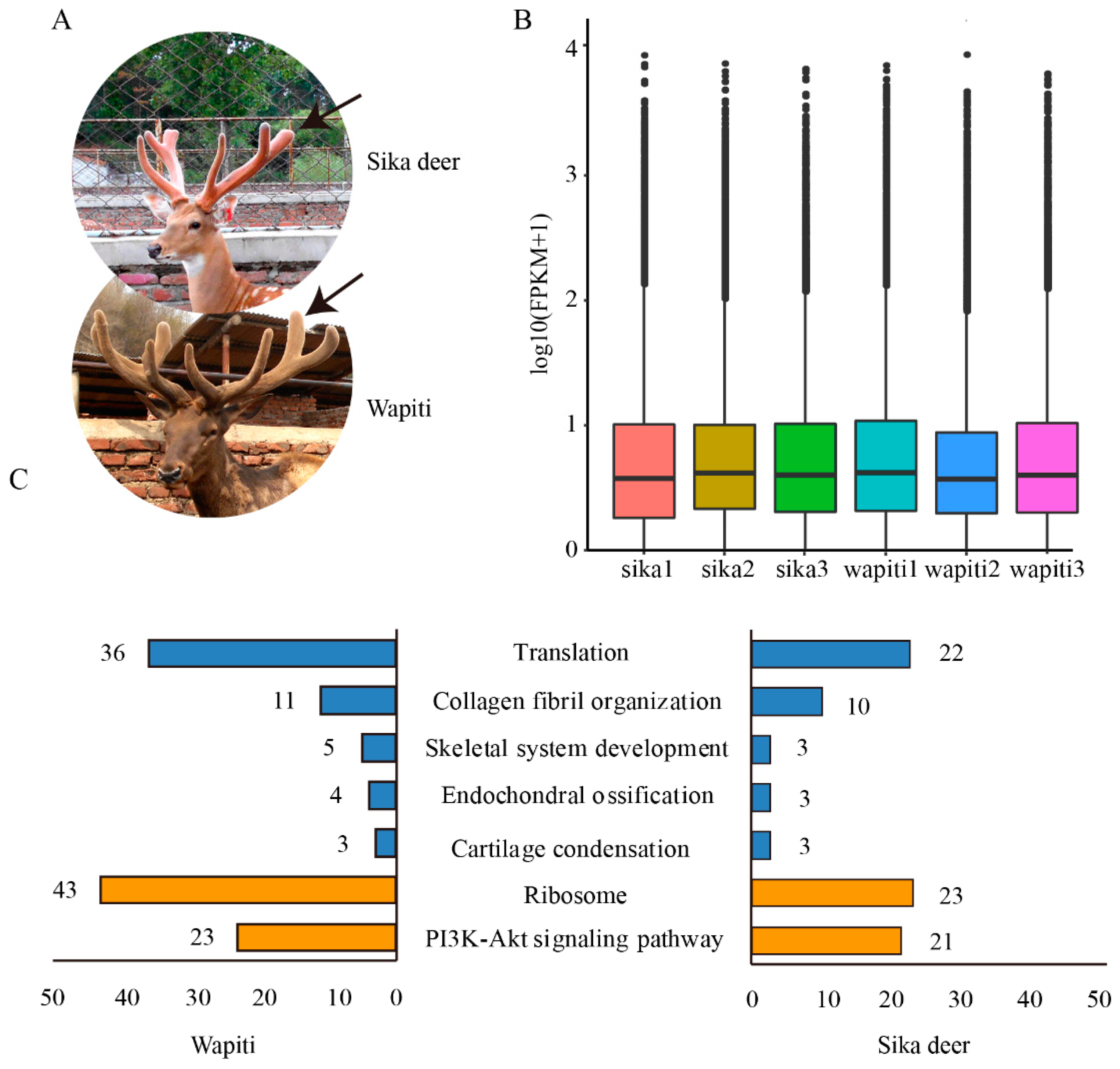
2.3. Reference Transcriptome Construction of Antlers
2.4. Analysis of Gene Expression in Wapiti and Sika Deer Antlers
2.5. Gene Function Enrichment Analysis
2.6. Orthologous Gene Analysis
2.7. Alternative Splicing Analysis
2.8. Gene Interaction Network Analysis
2.9. Gene Expression Profiling in Different Tissue Types from Antlers with Different Growth Rate
3. Results
3.1. Reference Transcriptome and Gene Expression Profiling of Antlers
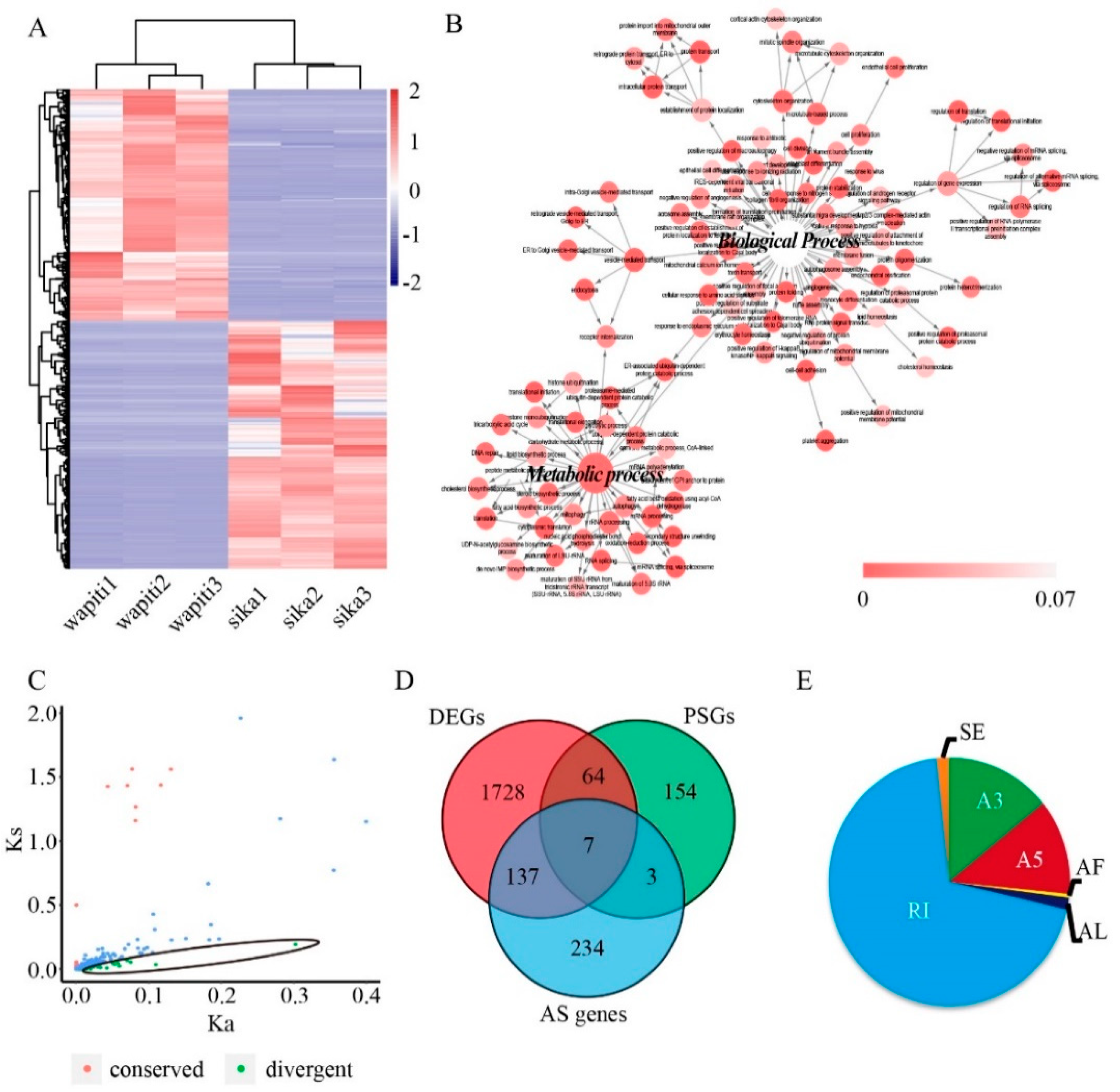
3.2. Characterization of Orthologous Genes of Wapiti and Sika Deer Antlers
3.3. Alternative Splicing Events in the Wapiti and Sika Deer Antlers
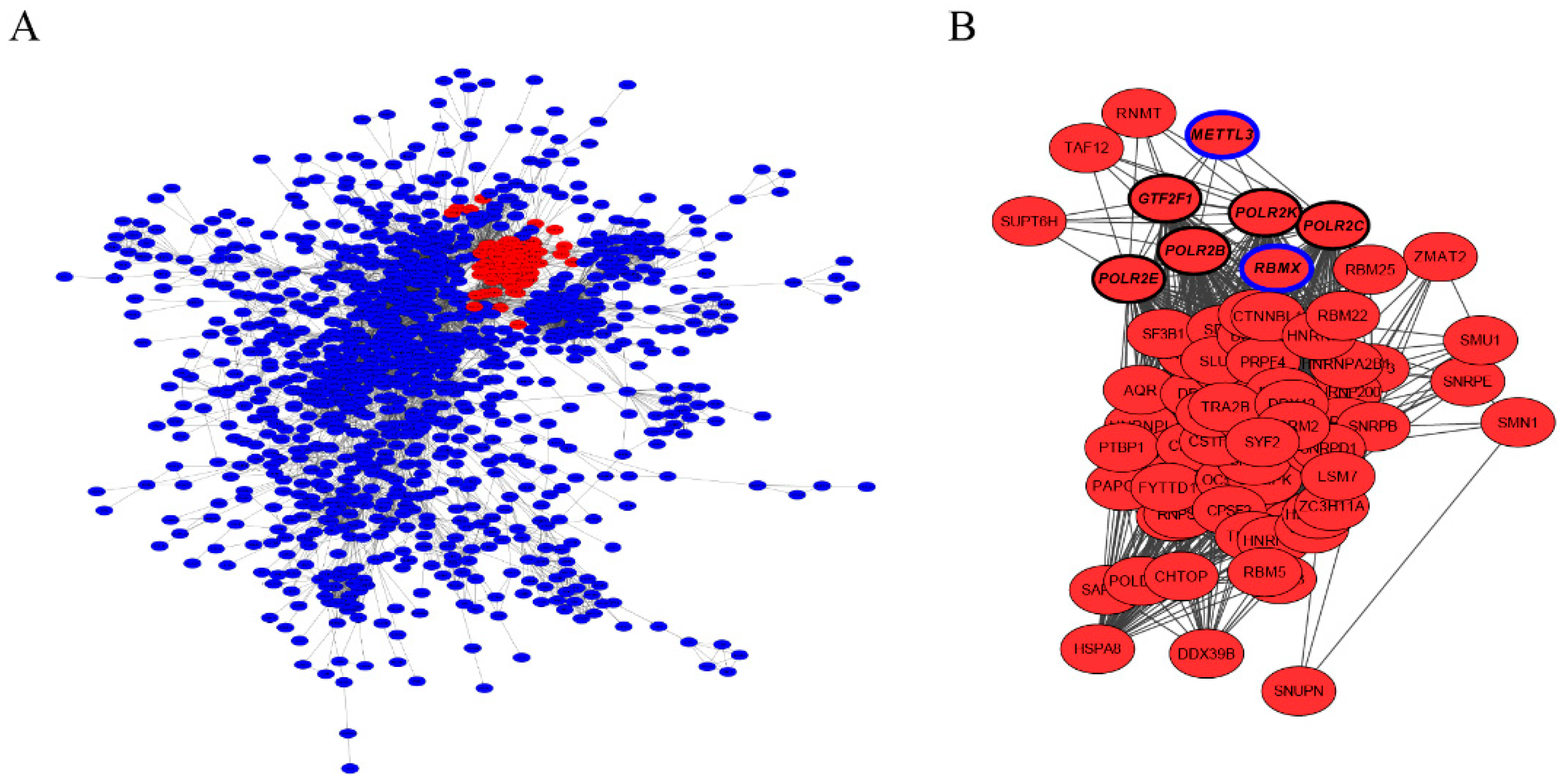
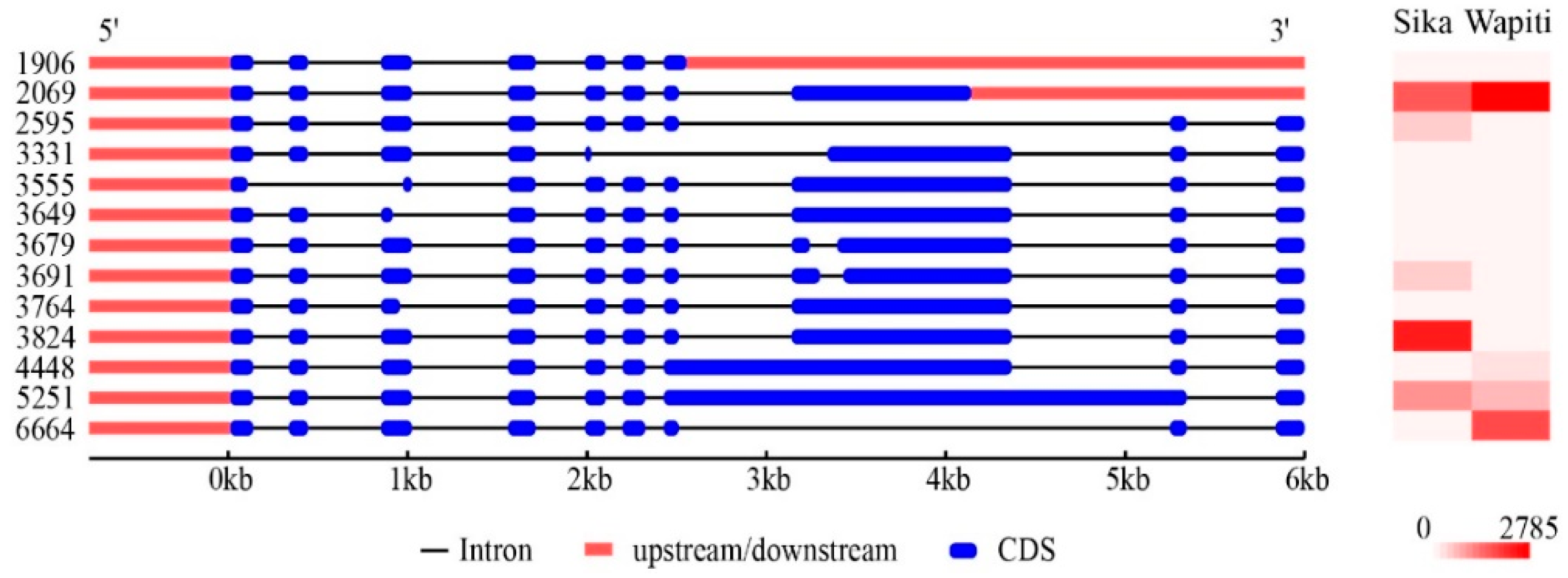
3.4. Characterization of Hub Genes Related with Regulation of Antler Growth Rate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goss, R.J. Deer Antlers: Regeneration, Function and Evolution; Academic Press: New York, NY, USA, 1983. [Google Scholar]
- Bartos, L.; Bahbouh, R. Antler size and fluctuating asymmetry in red deer (Cervus elaphus) stags and probability of becoming a harem holder in rut. Biol. J. Linn. Soc. 2006, 87, 59–68. [Google Scholar] [CrossRef]
- Liu, W.J. Remarks on Chinese materia medica—Young Pilose antler—A precious drug. Zhong Yao Tong Bao 1981, 6, 43–45. [Google Scholar] [PubMed]
- Li, C.; Suttie, J.M.; Clark, D.E. Histological examination of antler regeneration in red deer (Cervus elaphus). Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2005, 282, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E.; Li, C.Y.; Wang, W.Y.; Martin, S.K.; Suttie, J.M. Vascular localization and proliferation in the growing tip of the deer antler. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2006, 288A, 973–981. [Google Scholar] [CrossRef]
- Li, C.Y.; Yang, F.H.; Sheppard, A. Adult Stem Cells and Mammalian Epimorphic Regeneration—Insights from Studying Annual Renewal of Deer Antlers. Curr. Stem Cell Res. Ther. 2009, 4, 237–251. [Google Scholar] [CrossRef]
- Newbrey, J.W.; Banks, W.J. Ultrastructural changes associated with the mineralization of deer antler cartilage. Am. J. Anat. 1983, 166, 1–17. [Google Scholar] [CrossRef]
- Bubenik, G.A.; Sempere, A.J.; Hamr, J. Developing antler, a model for endocrine regulation of bone growth. Concentration gradient of T3, T4, and alkaline phosphatase in the antler, jugular, and the saphenous veins. Calcif. Tissue Int. 1987, 41, 38–43. [Google Scholar] [CrossRef]
- Li, C.; Clark, D.E.; Lord, E.A.; Stanton, J.A.; Suttie, J.M. Sampling technique to discriminate the different tissue layers of growing antler tips for gene discovery. Anat. Rec. 2002, 268, 125–130. [Google Scholar] [CrossRef]
- Li, C. Histogenetic aspects of deer antler development. Front. Biosci. 2013, 5, 479–489. [Google Scholar] [CrossRef]
- Hu, P.; Wang, T.; Liu, H.; Xu, J.; Wang, L.; Zhao, P.; Xing, X. Full-length transcriptome and microRNA sequencing reveal the specific gene-regulation network of velvet antler in sika deer with extremely different velvet antler weight. Mol. Genet. Genom. 2019, 294, 431–443. [Google Scholar] [CrossRef]
- Sadighi, M.; Haines, S.R.; Skottner, A.; Harris, A.J.; Suttie, J.M. Effects of insulin-like growth factor-I (IGF-I) and IGF-II on the growth of antler cells in vitro. J. Endocrinol. 1994, 143, 461–469. [Google Scholar] [CrossRef]
- Bielli, A.; Freitas-de-Melo, A.; Genovese, P.; Villagrán, M.; Tasende, C.; Ungerfeld, R. Androgen and oestrogen receptors in the growing antlers velvet of adult and yearling pampas deer (Ozotoceros bezoarticus) males. Anat. Histol. Embryol. 2021, 50, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, C.; Horton, M.A.; Price, J.S. Nuclear localization of type I parathyroid hormone/parathyroid hormone-related protein receptors in deer antler osteoclasts: Evidence for parathyroid hormone-related protein and receptor activator of NF-kappaB-dependent effects on osteoclast formation in regenerating mammalian bone. J. Bone Miner. Res. 2002, 17, 455–464. [Google Scholar] [PubMed]
- Feleke, M.; Bennett, S.; Chen, J.; Hu, X.; Williams, D.; Xu, J. New physiological insights into the phenomena of deer antler: A unique model for skeletal tissue regeneration. J. Orthop. Transl. 2020, 27, 57–66. [Google Scholar] [CrossRef]
- Steger, V.; Molnar, A.; Borsy, A.; Gyurjan, I.; Szabolcsi, Z.; Dancs, G.; Molnar, J.; Papp, P.; Nagy, J.; Puskas, L.; et al. Antler development and coupled osteoporosis in the skeleton of red deer Cervus elaphus: Expression dynamics for regulatory and effector genes. Mol. Genet. Genom. 2010, 284, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Bilezikian, J.P.; Raisz, L.G.; Martin, T.J. Principles of Bone Biology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2008. [Google Scholar]
- Molnár, A.; Gyurján, I.; Korpos, E.; Borsy, A.; Stéger, V.; Buzás, Z.; Kiss, I.; Zomborszky, Z.; Papp, P.; Deák, F.; et al. Identification of differentially expressed genes in the developing antler of red deer Cervus elaphus. Mol. Genet. Genom. 2007, 277, 237–248. [Google Scholar] [CrossRef]
- Gu, L.; Mo, E.; Sun, B.; Sung, C.; Jia, X.; Zhu, X.; Fang, Z. Analysis of gene expression in four parts of the red-deer antler using DNA chip microarray technology. Anim. Biol. 2008, 58, 67–90. [Google Scholar] [CrossRef]
- Pita-Thomas, W.; Fernández-Martos, C.; Yunta, M.; Maza, R.M.; Navarro-Ruiz, R.; Lopez-Rodríguez, M.J.; Reigada, D.; Nieto-Sampedro, M.; Nieto-Diaz, M. Gene expression of axon growth promoting factors in the deer antler. PLoS ONE 2010, 5, 0015706. [Google Scholar] [CrossRef]
- Hao, L.; Li, H.P.; Yan, L. Analysis of expressed sequence tags (ESTs) in sika deer (Cervus nippon hortulorum) velvet tip tissue. Yi Chuan 2011, 33, 371–377. [Google Scholar] [CrossRef]
- Yao, B.J.; Zhao, Y.; Zhang, H.S.; Zhang, M.; Liu, M.C.; Liu, H.L.; Li, J. Sequencing and de novo analysis of the Chinese Sika deer antler-tip transcriptome during the ossification stage using Illumina RNA-Seq technology. Biotechnol. Lett. 2012, 34, 813–822. [Google Scholar] [CrossRef]
- Zhao, Y.; Yao, B.J.; Zhang, M.; Wang, S.M.; Zhang, H.; Xiao, W. Comparative analysis of differentially expressed genes in Sika deer antler at different stages. Mol. Biol. Rep. 2013, 40, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Shao, Y.; Xu, J.; Wang, T.; Li, Y.; Liu, H.; Rong, M.; Su, W.; Chen, B.; Cui, S.; et al. Genome-wide study on genetic diversity and phylogeny of five species in the genus Cervus. BMC Genom. 2019, 20, 384. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Zhao, Y.; Wang, Q.; Zhang, M.; Liu, M.; Liu, H.; Li, J. De novo characterization of the antler tip of Chinese Sika deer transcriptome and analysis of gene expression related to rapid growth. Mol. Cell. Biochem. 2012, 364, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Ba, H.; Wang, D.; Yau, T.O.; Shang, Y.; Li, C. Transcriptomic analysis of different tissue layers in antler growth Center in Sika Deer (Cervus nippon). BMC Genom. 2019, 20, 173. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Zhang, M.; Liu, M.; Lu, B.; Leng, X.; Hu, Y.; Zhao, D.; Zhao, Y.U. Identification of the miRNA-mRNA regulatory network of antler growth centers. J. Biosci. 2019, 44, 11. [Google Scholar] [CrossRef]
- Kim, M.A.; Rhee, J.S.; Kim, T.H.; Lee, J.S.; Choi, A.Y.; Choi, B.S.; Choi, I.Y.; Sohn, Y.C. Alternative Splicing Profile and Sex-Preferential Gene Expression in the Female and Male Pacific Abalone Haliotis discus hannai. Genes 2017, 8, 99. [Google Scholar] [CrossRef]
- Zhang, S.J.; Wang, C.; Yan, S.; Fu, A.; Luan, X.; Li, Y.; Sunny Shen, Q.; Zhong, X.; Chen, J.Y.; Wang, X.; et al. Isoform Evolution in Primates through Independent Combination of Alternative RNA Processing Events. Mol. Biol. Evol. 2017, 34, 2453–2468. [Google Scholar] [CrossRef]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L.; et al. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar] [CrossRef]
- Wu, Y.; Xie, L.; Wang, M.; Xiong, Q.; Guo, Y.; Liang, Y.; Li, J.; Sheng, R.; Deng, P.; Wang, Y.; et al. Mettl3-mediated m(6)A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat. Commun. 2018, 9, 018–06898. [Google Scholar] [CrossRef]
- Barling, P.M.; Liu, H.; Matich, J.; Mount, J.; Ka Wai Lai, A.; Ma, L.; Basford Nicholson, L.F. Expression of PTHrP and the PTH/PTHrP receptor in growing red deer antler. Cell Biol. Int. 2004, 28, 661–673. [Google Scholar] [CrossRef]
- Fan, Y.; Hanai, J.I.; Le, P.T.; Bi, R.; Maridas, D.; DeMambro, V.; Figueroa, C.A.; Kir, S.; Zhou, X.; Mannstadt, M.; et al. Parathyroid Hormone Directs Bone Marrow Mesenchymal Cell Fate. Cell Metab. 2017, 25, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Lahn, B.T.; Page, D.C. Four evolutionary strata on the human X chromosome. Science 1999, 286, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Dass, B.; Tardif, S.; Park, J.Y.; Tian, B.; Weitlauf, H.M.; Hess, R.A.; Carnes, K.; Griswold, M.D.; Small, C.L.; Macdonald, C.C. Loss of polyadenylation protein tauCstF-64 causes spermatogenic defects and male infertility. Proc. Natl. Acad. Sci. USA 2007, 104, 20374–20379. [Google Scholar] [CrossRef]
- Liu, D.; Brockman, J.M.; Dass, B.; Hutchins, L.N.; Singh, P.; McCarrey, J.R.; MacDonald, C.C.; Graber, J.H. Systematic variation in mRNA 3′-processing signals during mouse spermatogenesis. Nucleic Acids Res. 2007, 35, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Choi, E.A.; Weng, L.; Xie, X.; Wan, J.; Xing, Y.; Moresco, J.J.; Tu, P.G.; Yates, J.R., 3rd; Shi, Y. Overlapping and distinct functions of CstF64 and CstF64τ in mammalian mRNA 3′ processing. Rna 2013, 19, 1781–1790. [Google Scholar] [CrossRef]
- Elliott, D.J.; Dalgliesh, C.; Hysenaj, G.; Ehrmann, I. RBMX family proteins connect the fields of nuclear RNA processing, disease and sex chromosome biology. Int. J. Biochem. Cell Biol. 2019, 108, 1–6. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Wang, N.; Li, Z.; Heller, R.; Liu, R.; Zhao, Y.; Han, J.; Pan, X.; Zheng, Z.; et al. Genetic basis of ruminant headgear and rapid antler regeneration. Science 2019, 364, eaav6335. [Google Scholar] [CrossRef]
- Tsend-Ayush, E.; O’Sullivan, L.A.; Grützner, F.S.; Onnebo, S.M.; Lewis, R.S.; Delbridge, M.L.; Marshall Graves, J.A.; Ward, A.C. RBMX gene is essential for brain development in zebrafish. Dev. Dyn. 2005, 234, 682–688. [Google Scholar] [CrossRef]
- Buchser, W.J.; Smith, R.P.; Pardinas, J.R.; Haddox, C.L.; Hutson, T.; Moon, L.; Hoffman, S.R.; Bixby, J.L.; Lemmon, V.P. Peripheral nervous system genes expressed in central neurons induce growth on inhibitory substrates. PLoS ONE 2012, 7, 6. [Google Scholar] [CrossRef]
- Zhang, J.; Li, D.; Shen, A.; Mao, H.; Jin, H.; Huang, W.; Xu, D.; Fan, J.; Chen, J.; Yang, L.; et al. Expression of RBMX after spinal cord injury in rats. J. Mol. Neurosci. 2013, 49, 417–429. [Google Scholar] [CrossRef]
- Climente-González, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Renieri, A.; Mencarelli, M.A.; Cetta, F.; Baldassarri, M.; Mari, F.; Furini, S.; Piu, P.; Ariani, F.; Dragani, T.A.; Frullanti, E. Oligogenic germline mutations identified in early non-smokers lung adenocarcinoma patients. Lung Cancer 2014, 85, 168–174. [Google Scholar] [CrossRef]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Munschauer, M.; Nguyen, C.T.; Sirokman, K.; Hartigan, C.R.; Hogstrom, L.; Engreitz, J.M.; Ulirsch, J.C.; Fulco, C.P.; Subramanian, V.; Chen, J.; et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature 2018, 561, 132–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Samples | Characteristic | Estimated Growth Rate of Antler | |
|---|---|---|---|---|
| Live deer | ||||
| Sika deer | 3 | Tip of main beam (MB) of antler (Figure 1A) | Growth rate | 93 ± 5 g/d |
| Sika deer | 3 | Growth rate | 47 ± 3 g/d | |
| Wapiti | 3 | Growth rate | 137 ± 8 g/d | |
| Slaughtered deer | ||||
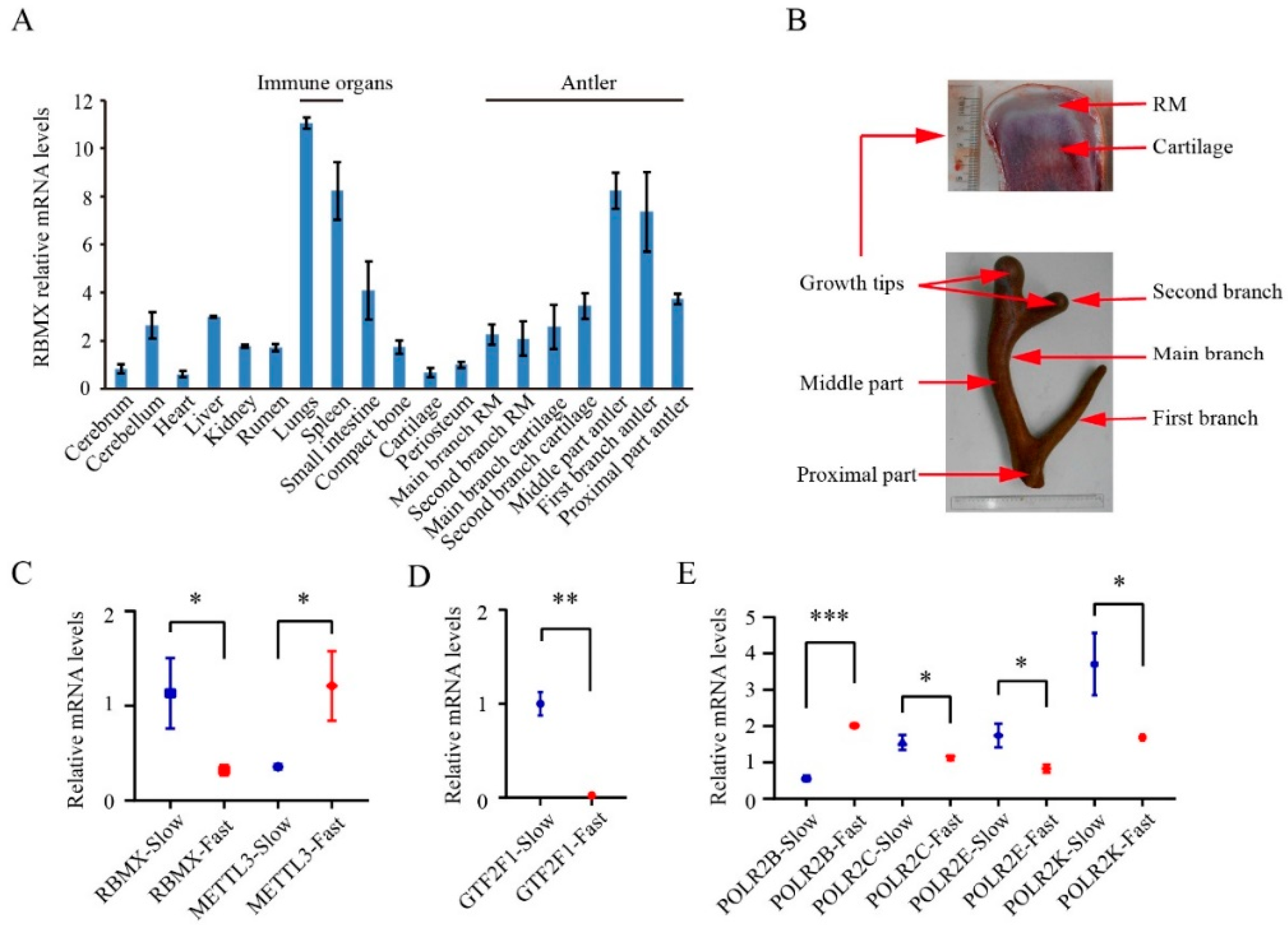
| Sika deer | 3 | Antler: MB growth tip cartilage; second branch (SB) growth tip RM and cartilage; middle part of antler; first branch antler; proximal part of the antler | ||
| Other tissues: cerebrum, cerebellum, heart, liver, kidney, rumen, lungs, spleen, small intestine, compact bone, cartilage and periosteum | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, P.; Wang, Z.; Li, J.; Wang, D.; Wang, Y.; Zhao, Q.; Li, C. Identification and Characterization of Alternative Splicing Variants and Positive Selection Genes Related to Distinct Growth Rates of Antlers Using Comparative Transcriptome Sequencing. Animals 2022, 12, 2203. https://doi.org/10.3390/ani12172203
Hu P, Wang Z, Li J, Wang D, Wang Y, Zhao Q, Li C. Identification and Characterization of Alternative Splicing Variants and Positive Selection Genes Related to Distinct Growth Rates of Antlers Using Comparative Transcriptome Sequencing. Animals. 2022; 12(17):2203. https://doi.org/10.3390/ani12172203
Chicago/Turabian StyleHu, Pengfei, Zhen Wang, Jiping Li, Dongxu Wang, Yusu Wang, Quanmin Zhao, and Chunyi Li. 2022. "Identification and Characterization of Alternative Splicing Variants and Positive Selection Genes Related to Distinct Growth Rates of Antlers Using Comparative Transcriptome Sequencing" Animals 12, no. 17: 2203. https://doi.org/10.3390/ani12172203