
Genome-Wide Identification and Characterization of Circular RNAs during Skeletal Muscle Development in Meat Rabbits

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Sample Preparation
2.3. Whole-Transcriptome Sequencing and Differential Analysis of mRNAs
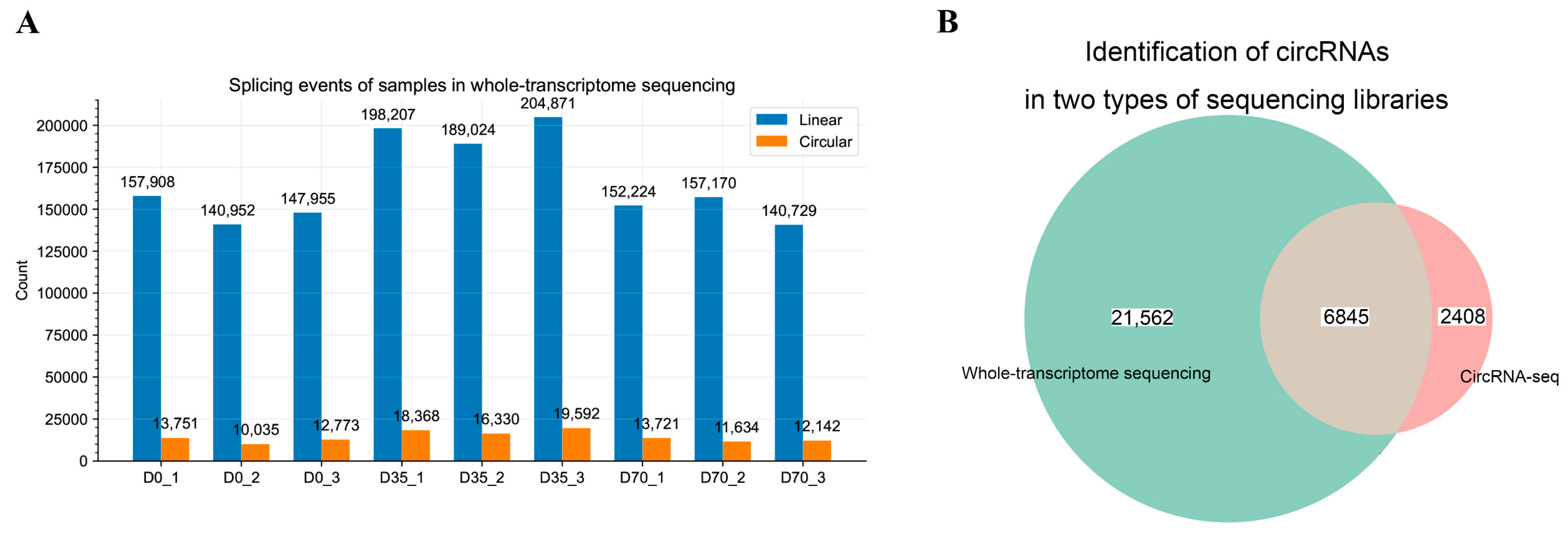
2.4. Identification of circRNAs from Whole-Transcriptome Sequencing Data
2.5. Identification and Assembly of Spliced Sequences of circRNA Using circRNA-seq
2.6. Differential Analysis of circRNAs
2.7. CircRNA-miRNA-mRNA Network Construction
2.8. Functional Annotation
2.9. Statistical Analysis
3. Results
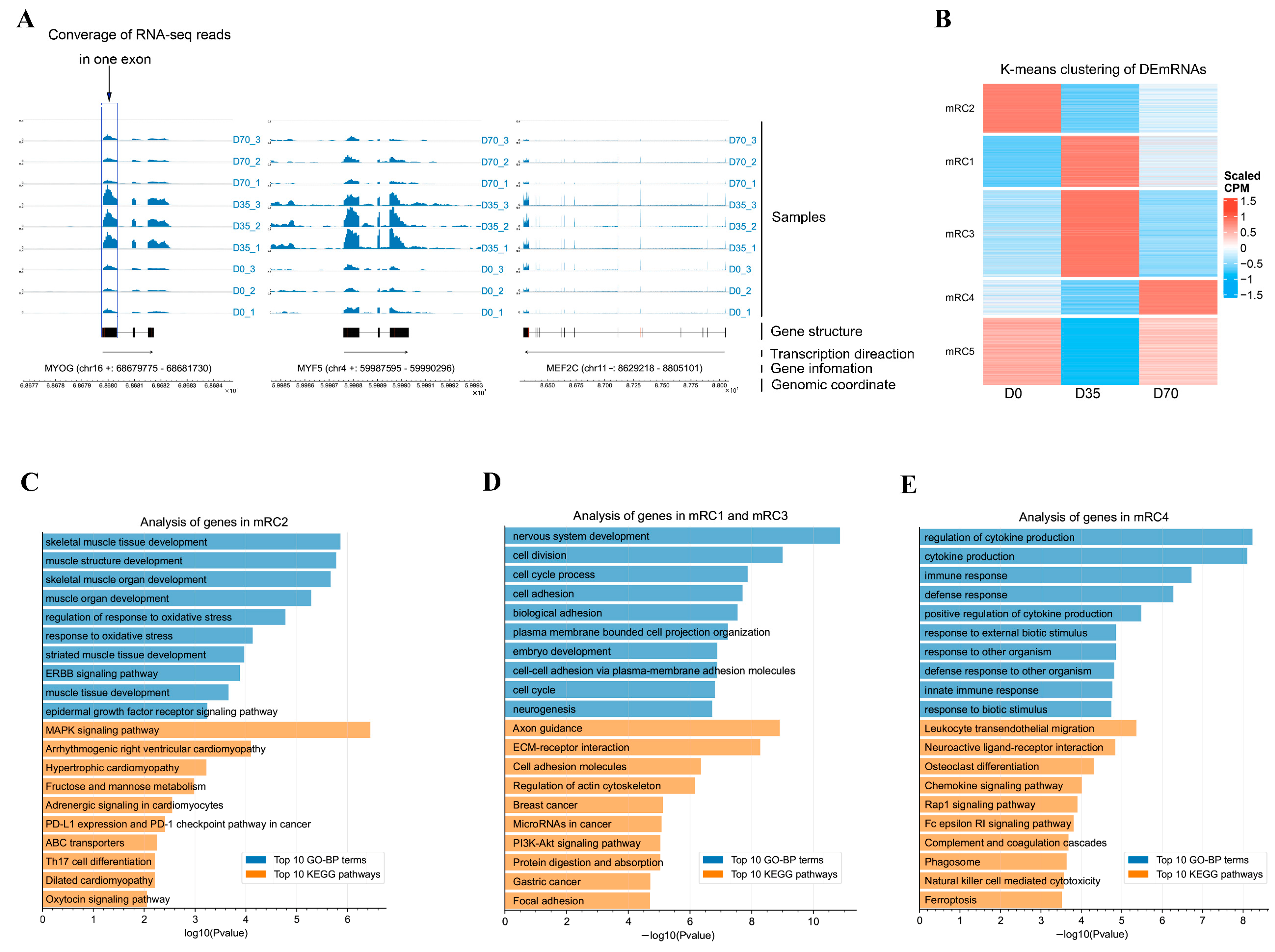
3.1. Differential Analysis of mRNAs during Skeletal Muscle Development in Rabbits
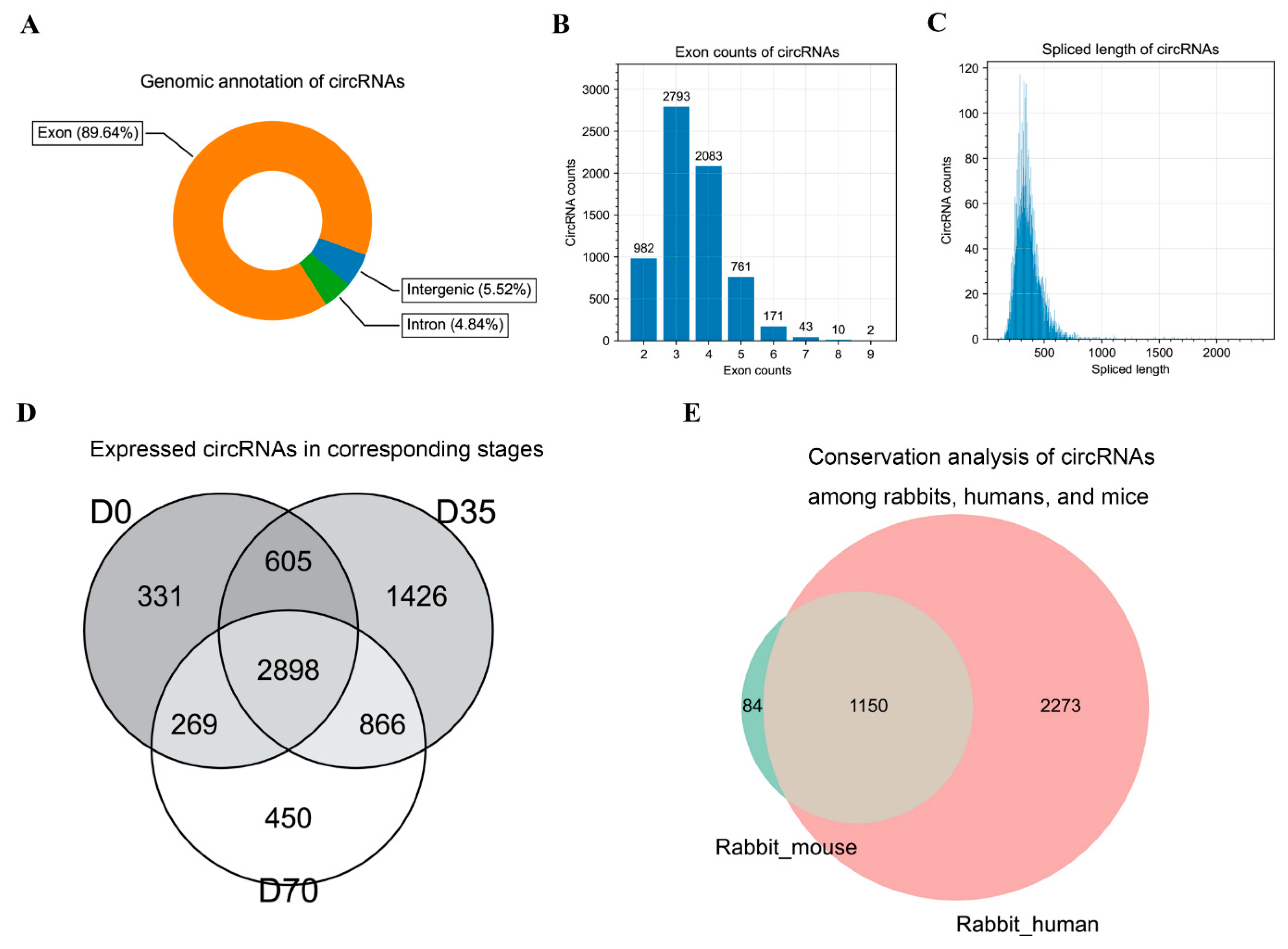
3.2. Identification and Characterization of circRNAs in Rabbit Skeletal Muscles
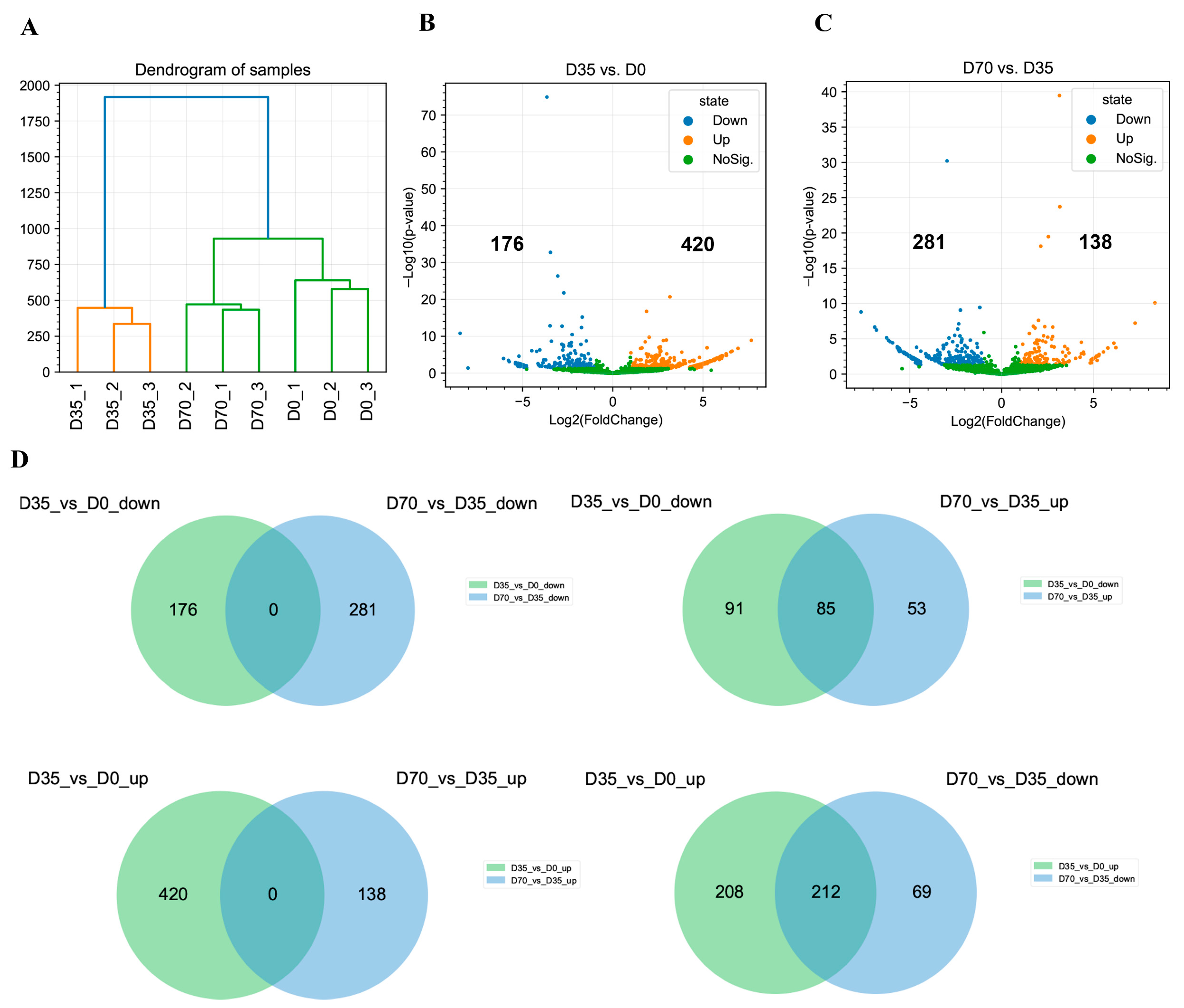
3.3. Differential Analysis of circRNAs during Skeletal Muscle Development in Rabbits
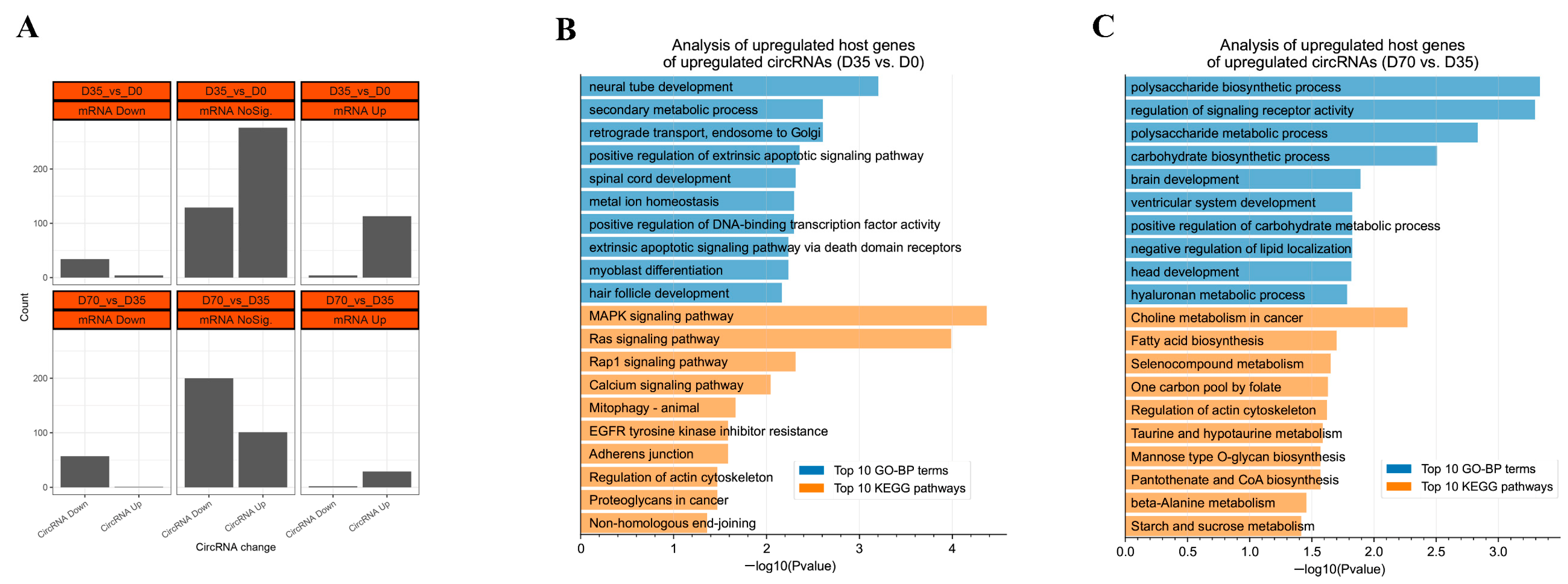
3.4. The Expression Relationships between circRNAs and Their Hose Genes
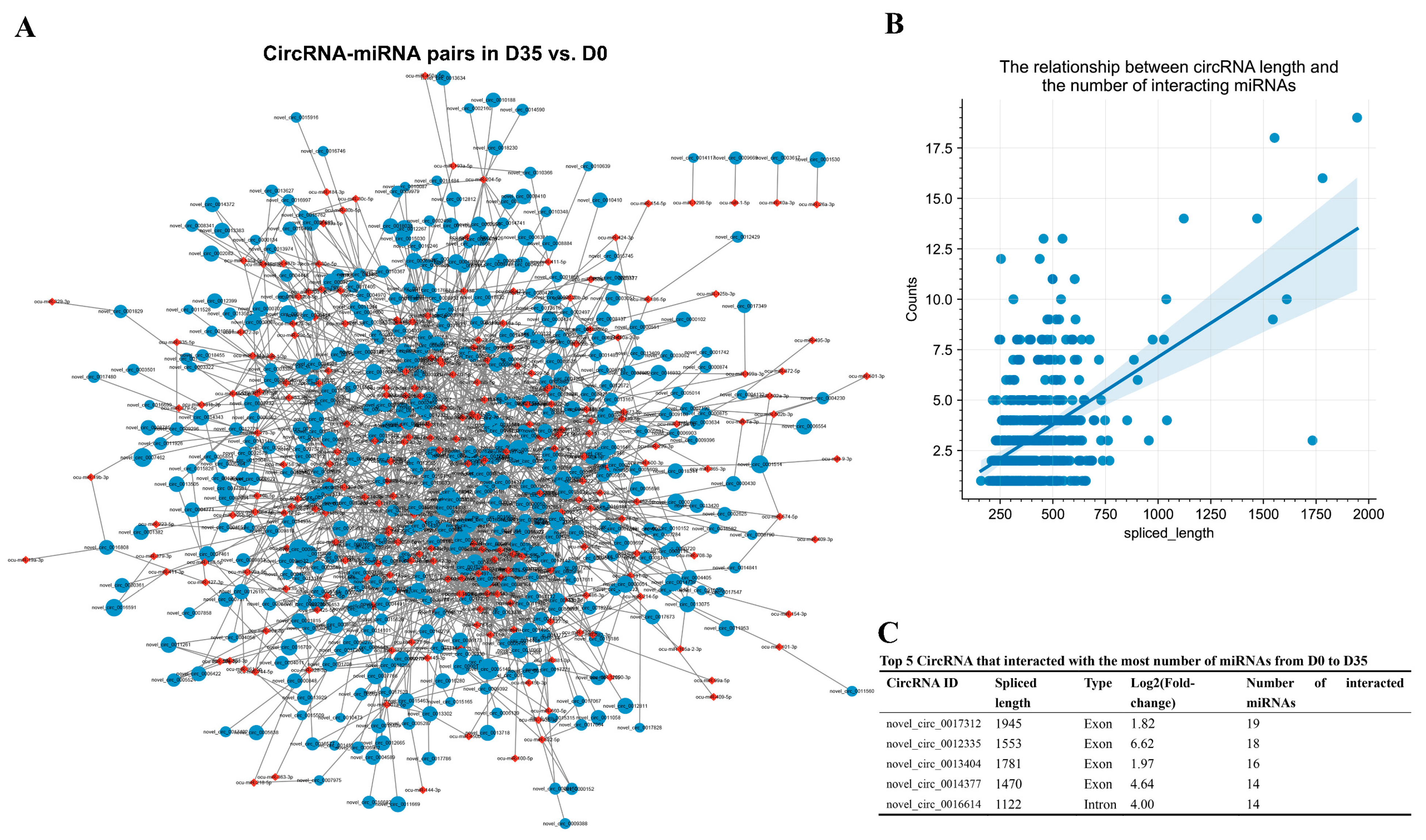
3.5. Construction of the DECs Involved Putative ceRNA Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, S.; Zeng, W.; Li, R.; Hoffman, L.C.; He, Z.; Sun, Q.; Li, H. Rabbit meat production and processing in China. Meat Sci. 2018, 145, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Madrid, B.S.; Hernández, P.; Blasco, A.; Haley, C.S.; Fontanesi, L.; Santacreu, M.A.; Pena, R.N.; Navarro, P.; Ibáñez-Escriche, N. Genomic regions influencing intramuscular fat in divergently selected rabbit lines. Anim. Genet. 2020, 51, 58–69. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Yvert, T.; Miyamoto-Mikami, E.; Tobina, T.; Shiose, K.; Kakigi, R.; Tsuzuki, T.; Takaragawa, M.; Ichinoseki-Sekine, N.; Pérez, M.; Kobayashi, H.; et al. PPARGC1A rs8192678 and NRF1 rs6949152 Polymorphisms Are Associated with Muscle Fiber Composition in Women. Genes 2020, 11, 1012. [Google Scholar] [CrossRef] [PubMed]
- Horak, M.; Novak, J.; Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Patop, I.L.; Wüst, S.; Kadener, S. Past, present, and future of circRNAs. EMBO J. 2019, 38, e100836. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ouyang, Z.; Shen, Y.; Liu, B.; Zhang, Q.; Wan, L.; Yin, Z.; Zhu, W.; Li, S.; Peng, D. CircRNA_28313/miR-195a/CSF1 axis modulates osteoclast differentiation to affect OVX-induced bone absorption in mice. RNA Biol. 2019, 16, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Chen, Y.G. Circular RNAs in Immune Response and Viral Infection. Trends Biochem. Sci. 2020, 45, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Zheng, G.; Ning, Q.; Zheng, J.; Dong, D. Translation and functional roles of circular RNAs in human cancer. Mol. Cancer 2020, 19, 30. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhang, J.; Tian, Y.; Gao, Y.; Dong, X.; Chen, W.; Yuan, X.; Yin, W.; Xu, J.; Chen, K.; et al. CircRNA inhibits DNA damage repair by interacting with host gene. Mol. Cancer 2020, 19, 128. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Chen, Z.; He, Z.; Xu, Y.; Li, Z. CircRNA-ENO1 promoted glycolysis and tumor progression in lung adenocarcinoma through upregulating its host gene ENO1. Cell Death Dis. 2019, 10, 885. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Yang, Y.; Niu, G.; Tang, Z.; Li, K. Genome-wide profiling of Sus scrofa circular RNAs across nine organs and three developmental stages. DNA Res. 2017, 24, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ren, Q.; Hua, L.; Chen, J.; Zhang, J.; Bai, H.; Li, H.; Xu, B.; Shi, Z.; Cao, H.; et al. Comprehensive Analysis of Differentially Expressed mRNA, lncRNA and circRNA and Their ceRNA Networks in the Longissimus Dorsi Muscle of Two Different Pig Breeds. Int. J. Mol. Sci. 2019, 20, 1107. [Google Scholar] [CrossRef]
- Ling, Y.; Zheng, Q.; Zhu, L.; Xu, L.; Sui, M.; Zhang, Y.; Liu, Y.; Fang, F.; Chu, M.; Ma, Y.; et al. Trend analysis of the role of circular RNA in goat skeletal muscle development. BMC Genom. 2020, 21, 220. [Google Scholar] [CrossRef]
- Shen, X.; Tang, J.; Ru, W.; Zhang, X.; Huang, Y.; Lei, C.; Cao, H.; Lan, X.; Chen, H. CircINSR Regulates Fetal Bovine Muscle and Fat Development. Front. Cell Dev. Biol. 2020, 8, 615638. [Google Scholar] [CrossRef] [PubMed]
- Hernández, P.; Cesari, V.; Blasco, A. Effect of genetic rabbit lines on lipid content, lipolytic activities and fatty acid composition of hind leg meat and perirenal fat. Meat Sci. 2008, 78, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; He, Z.; Lv, J.; Zhang, E.; Li, H. Identification the Key Odorants in Different Parts of Hyla Rabbit Meat via Solid Phase Microextraction Using Gas Chromatography Mass Spectrometry. Korean J. Food Sci. Anim. Resour. 2016, 36, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, C.; Yang, C.; Kuang, L.; Zheng, J.; Tang, L.; Lei, M.; Li, C.; Ren, Y.; Guo, Z.; et al. Circular RNA, microRNA and Protein Profiles of the Longissimus Dorsi of Germany ZIKA and Sichuan White Rabbits. Front. Genet. 2021, 12, 777232. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Guo, G.; Tian, X.; Hu, S.; Du, K.; Zhang, Q.; Mao, J.; Jia, X.; Chen, S.; Wang, J.; et al. Screening and identification of MicroRNAs expressed in perirenal adipose tissue during rabbit growth. Lipids Health Dis. 2020, 19, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018, 19, 803–810. [Google Scholar] [CrossRef]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011, 39, D38–D51. [Google Scholar] [CrossRef]
- Jing, J.; Jiang, X.; Zhu, C.; Zheng, Q.; Ji, Q.; Yin, H.; Huang, J.; Zhu, Y.; Wang, J.; Qin, S.; et al. Dynamic changes of miRNAs in skeletal muscle development at New Zealand rabbits. BMC Genom. 2021, 22, 577. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Riffo-Campos, Á.L.; Riquelme, I.; Brebi-Mieville, P. Tools for Sequence-Based miRNA Target Prediction: What to Choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Aase-Remedios, M.E.; Coll-Lladó, C.; Ferrier, D.E.K. More Than One-to-Four via 2R: Evidence of an Independent Amphioxus Expansion and Two-Gene Ancestral Vertebrate State for MyoD-Related Myogenic Regulatory Factors (MRFs). Mol. Biol. Evol. 2020, 37, 2966–2982. [Google Scholar] [CrossRef]
- Taylor, M.V.; Hughes, S.M. Mef2 and the skeletal muscle differentiation program. Semin. Cell Dev. Biol. 2017, 72, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Petrany, M.J.; Swoboda, C.O.; Sun, C.; Chetal, K.; Chen, X.; Weirauch, M.T.; Salomonis, N.; Millay, D.P. Single-nucleus RNA-seq identifies transcriptional heterogeneity in multinucleated skeletal myofibers. Nat. Commun. 2020, 11, 6374. [Google Scholar] [CrossRef]
- Xu, F.; Jiang, L.; Zhao, Q.; Zhang, Z.; Liu, Y.; Yang, S.; Yu, M.; Chen, H.; Zhang, J.; Zhang, J. Whole-transcriptome and proteome analyses identify key differentially expressed mRNAs, miRNAs, lncRNAs and circRNAs associated with HCC. Oncogene 2021, 40, 4820–4831. [Google Scholar] [CrossRef]
- Liu, X.; Bai, Y.; Cui, R.; He, S.; Zhao, X.; Wu, K.; Fang, M. Sus_circPAPPA2 Regulates Fat Deposition in Castrated Pigs through the miR-2366/GK Pathway. Biomolecules 2022, 12, 753. [Google Scholar] [CrossRef]
- Liu, R.; Liu, X.; Bai, X.; Xiao, C.; Dong, Y. Identification and Characterization of circRNA in Longissimus Dorsi of Different Breeds of Cattle. Front. Genet. 2020, 11, 565085. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, L.; Liu, X.; Niu, M.; Cui, J.; Che, S.; Liu, Y.; An, X.; Cao, B. Analyses of circRNA profiling during the development from pre-receptive to receptive phases in the goat endometrium. J. Anim. Sci. Biotechnol. 2019, 10, 34. [Google Scholar] [CrossRef]
- Varela-Martínez, E.; Corsi, G.I.; Anthon, C.; Gorodkin, J.; Jugo, B.M. Novel circRNA discovery in sheep shows evidence of high backsplice junction conservation. Sci. Rep. 2021, 11, 427. [Google Scholar] [CrossRef] [PubMed]
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Liu, W.; Liu, H.; Meng, Q.; Xu, Y.; Guo, Y.; Wang, B.; Zhou, Z.; Hou, S. Dynamic accumulation of fatty acids in duck (Anas platyrhynchos) breast muscle and its correlations with gene expression. BMC Genom. 2020, 21, 58. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Marics, I.; Padilla, F.; Guillemot, J.F.; Scaal, M.; Marcelle, C. FGFR4 signaling is a necessary step in limb muscle differentiation. Development 2002, 129, 4559–4569. [Google Scholar] [CrossRef]
- Soileau, L.C.; Silberstein, L.; Blau, H.M.; Thompson, W.J. Reinnervation of muscle fiber types in the newborn rat soleus. J. Neurosci. 1987, 7, 4176–4194. [Google Scholar] [CrossRef]
- Fernandes, J.J.; Keshishian, H. Nerve-muscle interactions during flight muscle development in Drosophila. Development 1998, 125, 1769–1779. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef]
- Merz, K.E.; Thurmond, D.C. Role of Skeletal Muscle in Insulin Resistance and Glucose Uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, K.; Zhao, X.; Li, Y.; Wu, Z.; Sun, W.; Wang, J.; Jia, X.; Chen, S.; Lai, S. Genome-Wide Identification and Characterization of Circular RNAs during Skeletal Muscle Development in Meat Rabbits. Animals 2022, 12, 2208. https://doi.org/10.3390/ani12172208
Du K, Zhao X, Li Y, Wu Z, Sun W, Wang J, Jia X, Chen S, Lai S. Genome-Wide Identification and Characterization of Circular RNAs during Skeletal Muscle Development in Meat Rabbits. Animals. 2022; 12(17):2208. https://doi.org/10.3390/ani12172208
Chicago/Turabian StyleDu, Kun, Xiaoyu Zhao, Yanhong Li, Zhoulin Wu, Wenqiang Sun, Jie Wang, Xianbo Jia, Shiyi Chen, and Songjia Lai. 2022. "Genome-Wide Identification and Characterization of Circular RNAs during Skeletal Muscle Development in Meat Rabbits" Animals 12, no. 17: 2208. https://doi.org/10.3390/ani12172208
APA StyleDu, K., Zhao, X., Li, Y., Wu, Z., Sun, W., Wang, J., Jia, X., Chen, S., & Lai, S. (2022). Genome-Wide Identification and Characterization of Circular RNAs during Skeletal Muscle Development in Meat Rabbits. Animals, 12(17), 2208. https://doi.org/10.3390/ani12172208