
Staphylococcus aureus-Induced Necroptosis Promotes Mitochondrial Damage in Goat Endometrial Epithelial Cells
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Bacterial Strains
2.2. Bacterial Infection
2.3. Lactate Dehydrogenase (LDH) Release
2.4. Cell Death Detection
2.5. Transmission Electron Microscopy (TEM)
2.6. Western Blot Analysis
2.7. Indirect Immunofluorescence Assay (IFA)
2.8. JC-1 Staining
2.9. Cytochrome C Assay
2.10. ROS Assay
2.11. Calcium Ion Imaging
2.12. Statistical Analysis
3. Results
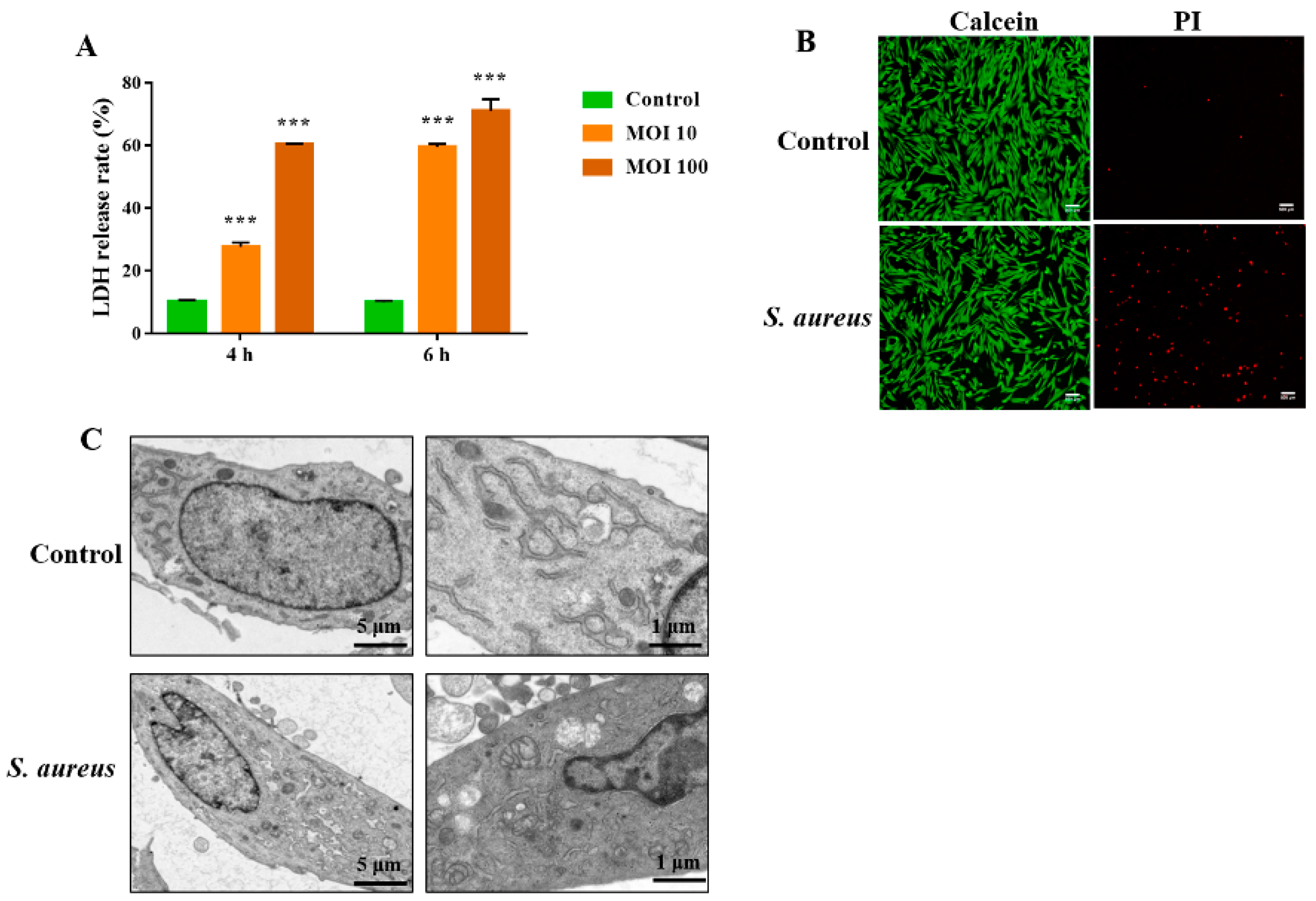
3.1. S. aureus Induces gEEC Necrotic Cell Death
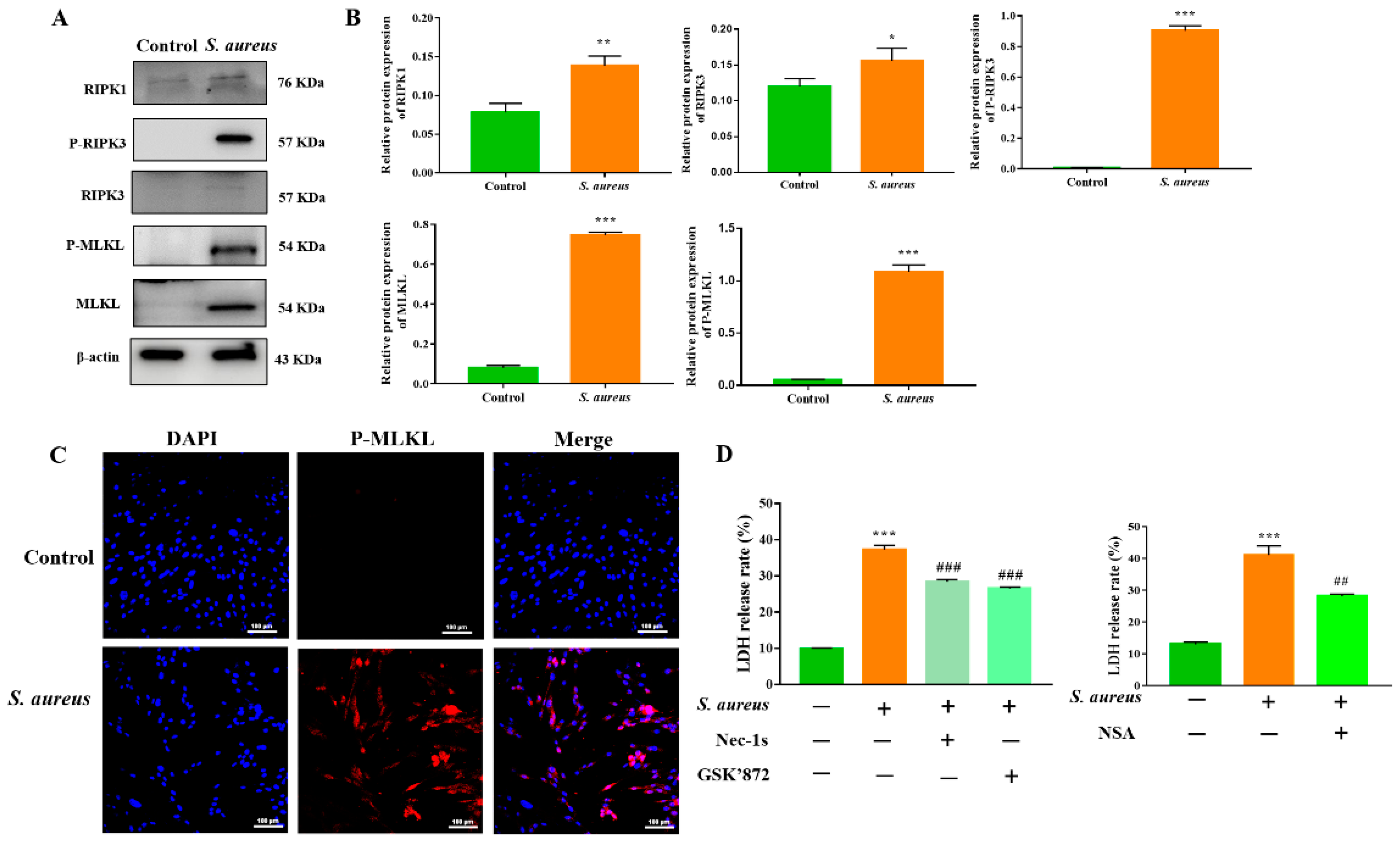
3.2. The RIPK1/RIPK3/MLKL Signaling Pathway Contributes to S. aureus-Induced gEEC Necroptosis
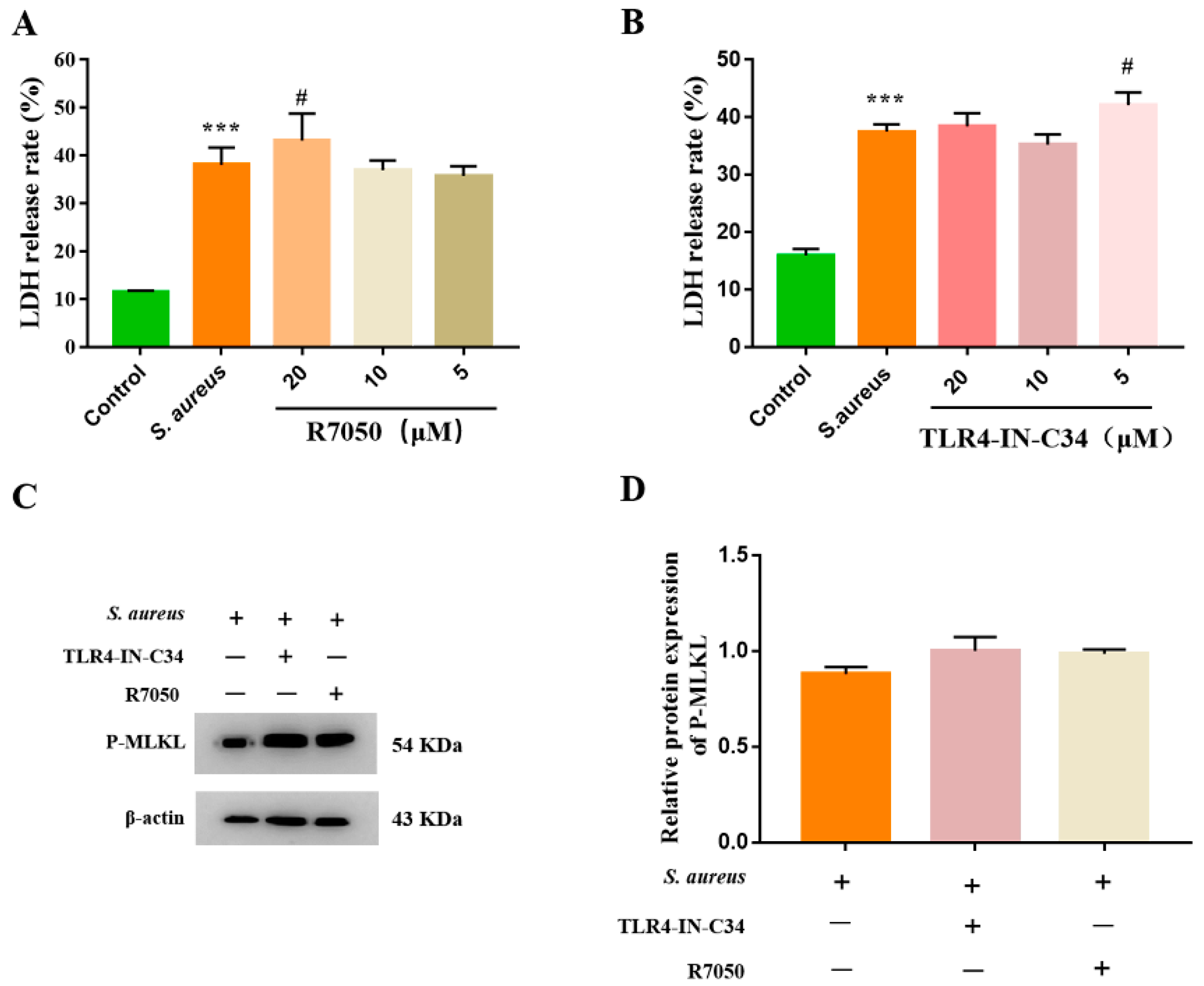
3.3. S. aureus-Induced gEEC Necroptosis Is Not Dependent on Death Receptor Signaling and TLR4
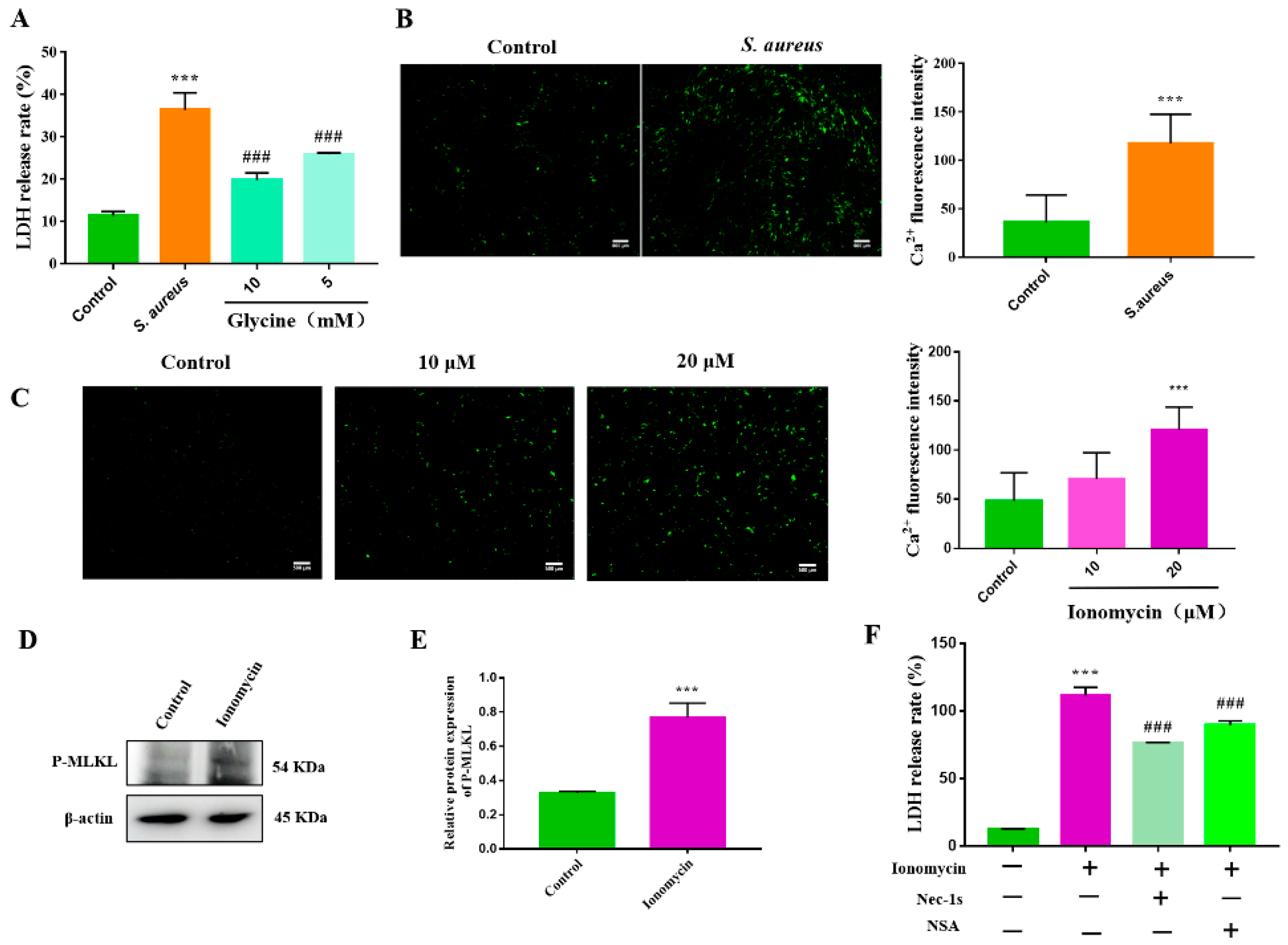
3.4. S. aureus-Induced gEEC Necroptosis Is Triggered by Ion Dysregulation and Membrane Disruption
3.5. Necroptosis Exacerbates the S. aureus-Induced gEEC Mitochondrial Damage and ROS Generation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carneiro, L.C.; Cronin, J.G.; Sheldon, I.M. Mechanisms linking bacterial infections of the bovine endometrium to disease and infertility. Reprod. Biol. 2016, 16, 1–7. [Google Scholar] [CrossRef]
- Gilbert, R.O.; Santos, N.R. Dynamics of postpartum endometrial cytology and bacteriology and their relationship to fertility in dairy cows. Theriogenology 2016, 85, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, I.M.; Price, S.B.; Cronin, J.; Gilbert, R.O.; Gadsby, J.E. Mechanisms of infertility associated with clinical and subclinical endometritis in high producing dairy cattle. Reprod. Domest. Anim. 2009, 44, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, Z.A.; Mann, G.E.; Robinson, R.S. Impact of endometritis on post-partum ovarian cyclicity in dairy cows. Vet. J. 2019, 248, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.J.; Piersanti, R.L.; Ramirez-Hernandez, R.; de Oliveira, E.B.; Bishop, J.V.; Hansen, T.R.; Ma, Z.; Jeong, K.; Santos, J.; Sheldon, M.I.; et al. Experimentally induced endometritis impairs the developmental capacity of bovine oocytes. Biol. Reprod. 2020, 103, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, M.; Peralta, M.B.; Angeli, E.; Stassi, A.F.; Gareis, N.C.; Durante, L.; Cainelli, S.; Salvetti, N.R.; Rey, F.; Ortega, H.H. Immune status during postpartum, peri-implantation and early pregnancy in cattle: An updated view. Anim. Reprod. Sci. 2019, 206, 1–10. [Google Scholar] [CrossRef]
- Suleymanov, S.M.; Usha, B.V.; Vatnikov, Y.A.; Sotnikova, E.D.; Kulikov, E.V.; Parshina, V.I.; Bolshakova, M.V.; Lyshko, M.U.; Romanova, E.V. Structural uterine changes in postpartum endometritis in cows. Vet. World 2018, 11, 1473–1478. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and relevance to disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Imre, G. The involvement of regulated cell death forms in modulating the bacterial and viral pathogenesis. Int. Rev. Cell Mol. Biol. 2020, 353, 211–253. [Google Scholar] [PubMed]
- Kothari, H.; Keshava, S.; Vatsyayan, R.; Mackman, N.; Rao, L.V.; Pendurthi, U.R. Role of tissue factor in Mycobacterium tuberculosis-induced inflammation and disease pathogenesis. PLoS ONE 2014, 9, e114141. [Google Scholar]
- Morinaga, Y.; Yanagihara, K.; Nakamura, S.; Hasegawa, H.; Seki, M.; Izumikawa, K.; Kakeya, H.; Yamamoto, Y.; Yamada, Y.; Kohno, S.; et al. Legionella pneumophila induces cathepsin B-dependent necrotic cell death with releasing high mobility group box1 in macrophages. Respir. Res. 2010, 11, 158. [Google Scholar] [CrossRef]
- Chen, F.; He, Y. Caspase-2 mediated apoptotic and necrotic murine macrophage cell death induced by rough Brucella abortus. PLoS ONE 2009, 4, e6830. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.; McComb, S.; Mulligan, R.; Dudani, R.; Krishnan, L.; Sad, S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 2012, 13, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.; Cohen, T.S.; Chung, S.; Wachtel, S.; Bueno, S.; Prince, A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015, 11, e1004820. [Google Scholar] [CrossRef]
- Kitur, K.; Wachtel, S.; Brown, A.; Wickersham, M.; Paulino, F.; Peñaloza, H.F.; Soong, G.; Bueno, S.; Parker, D.; Prince, A. Necroptosis promotes Staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep. 2016, 16, 2219–2230. [Google Scholar] [CrossRef] [PubMed]
- González-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PLoS Pathog. 2015, 11, e1005337. [Google Scholar] [CrossRef]
- González-Juarbe, N.; Bradley, K.M.; Shenoy, A.T.; Gilley, R.P.; Reyes, L.F.; Hinojosa, C.A.; Restrepo, M.I.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 2017, 24, 917–928. [Google Scholar] [CrossRef]
- Li, B.; Yang, N.; Shan, Y.; Wang, X.; Hao, Y.; Mao, R.; Teng, D.; Fan, H.; Wang, J. Therapeutic potential of a designed CSαβ peptide ID13 in Staphylococcus aureus-induced endometritis of mice. Appl. Microbiol. Biotechnol. 2020, 104, 6693–6705. [Google Scholar] [CrossRef]
- Wong Fok Lung, T.; Monk, I.R.; Acker, K.P.; Mu, A.; Wang, N.; Riquelme, S.A.; Pires, S.; Noguera, L.P.; Dach, F.; Gabryszewski, S.J.; et al. Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat. Microbiol. 2020, 5, 141–153. [Google Scholar] [CrossRef]
- Zhou, Y.; Niu, C.; Ma, B.; Xue, X.; Li, Z.; Chen, Z.; Li, F.; Zhou, S.; Luo, X.; Hou, Z. Inhibiting PSMα-induced neutrophil necroptosis protects mice with MRSA pneumonia by blocking the agr system. Cell Death Dis. 2018, 9, 362. [Google Scholar] [CrossRef] [Green Version]
- Tam, K.; Torres, V.J. Staphylococcus aureus secreted toxins and extracellular enzymes. Microbiol. Spectr. 2019, 7, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Seilie, E.S.; Bubeck Wardenburg, J. Staphylococcus aureus pore-forming toxins: The interface of pathogen and host complexity. Semin. Cell Dev. Biol. 2017, 72, 101–116. [Google Scholar] [CrossRef]
- Ioannidi, K.S.; Vasileiou, N.; Barbagianni, M.S.; Orfanou, D.C.; Mantziaras, G.; Chouzouris, T.M.; Dovolou, E.; Chatzopoulos, D.C.; Karavanis, E.; Papadopoulos, N.; et al. Clinical, ultrasonographic, bacteriological, cytological and histopathological findings of uterine involution in ewes with uterine infection. Pathogens 2020, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.B.; Sui, G.G.; Lu, X.Y.; Sun, Z.L. Elevated levels of ADMA are associated with lower DDAH2 and higher PRMT1 in LPS-induced endometritis rats. Inflammation 2018, 41, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol. Immunol. 2020, 64, 252–269. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; Fitzgibbon, C.; et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 2020, 11, 3151. [Google Scholar] [CrossRef]
- Davies, D.; Meade, K.G.; Herath, S.; Eckersall, P.D.; Gonzalez, D.; White, J.O.; Conlan, R.S.; O’Farrelly, C.; Sheldon, I.M. Toll-like receptor and antimicrobial peptide expression in the bovine endometrium. Reprod. Biol. Endocrinol. 2008, 6, 53. [Google Scholar] [CrossRef]
- Xia, X.; Lei, L.; Wang, S.; Hu, J.; Zhang, G. Necroptosis and its role in infectious diseases. Apoptosis 2020, 25, 169–178. [Google Scholar] [CrossRef]
- Samson, A.L.; Garnish, S.E.; Hildebrand, J.M.; Murphy, J.M. Location, location, location: A compartmentalized view of TNF-induced necroptotic signaling. Sci. Signal. 2021, 14, eabc6178. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef] [PubMed]
- Roca, F.J.; Ramakrishnan, L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013, 153, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef]
- Seol, J.W.; Kang, S.J.; Park, S.Y. Silver ion treatment of primary cultured bovine mammary gland epithelial cell (BMEC) damage from Staphylococcus aureus-derived alpha-toxin. Vet. Res. Commun. 2010, 34, 33–42. [Google Scholar] [CrossRef]
- Lee, K.I.; Choi, S.; Choi, H.G.; Gurmessa, S.K.; Dang, T.B.; Back, Y.W.; Park, H.S.; Kim, H.J. Recombinant Rv1654 protein of Mycobacterium tuberculosis induces mitochondria-mediated apoptosis in macrophage. Microbiol. Immunol. 2021, 65, 178–188. [Google Scholar] [CrossRef]
- Shaukat, A.; Shaukat, I.; Rajput, S.A.; Shukat, R.; Hanif, S.; Jiang, K.; Zhang, T.; Akhtar, M.; Shaukat, I.; Ma, X.; et al. Ginsenoside Rb1 protects from Staphylococcus aureus-induced oxidative damage and apoptosis through endoplasmic reticulum-stress and death receptor-mediated pathways. Ecotoxicol. Environ. Saf. 2021, 219, 112353. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef]
- Basit, F.; van Oppen, L.M.; Schöckel, L.; Bossenbroek, H.M.; van Emst-de Vries, S.E.; Hermeling, J.C.; Grefte, S.; Kopitz, C.; Heroult, M.; Hgm Willems, P.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, M.; Wei, R.; Wang, J.; Qin, J.; Yu, D.; Guan, X.; Li, X.; Fu, M.; Liu, H.; Chen, X. Osthole induces necroptosis via ROS overproduction in glioma cells. FEBS Open Bio 2021, 11, 456–467. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, Y.; Gao, K.; Lin, P.; Chen, H.; Zhou, D.; Tang, K.; Wang, A.; Jin, Y. Staphylococcus aureus-Induced Necroptosis Promotes Mitochondrial Damage in Goat Endometrial Epithelial Cells. Animals 2022, 12, 2218. https://doi.org/10.3390/ani12172218
Yi Y, Gao K, Lin P, Chen H, Zhou D, Tang K, Wang A, Jin Y. Staphylococcus aureus-Induced Necroptosis Promotes Mitochondrial Damage in Goat Endometrial Epithelial Cells. Animals. 2022; 12(17):2218. https://doi.org/10.3390/ani12172218
Chicago/Turabian StyleYi, Yanyan, Kangkang Gao, Pengfei Lin, Huatao Chen, Dong Zhou, Keqiong Tang, Aihua Wang, and Yaping Jin. 2022. "Staphylococcus aureus-Induced Necroptosis Promotes Mitochondrial Damage in Goat Endometrial Epithelial Cells" Animals 12, no. 17: 2218. https://doi.org/10.3390/ani12172218