
Genome-Wide Population Structure Analysis and Genetic Diversity Detection of Four Chinese Indigenous Duck Breeds from Fujian Province

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Samples and DNA Extraction
2.2. RAD Sequencing and Data Sorting
2.3. Population Structure Analysis
2.4. ROH Analysis
3. Results
3.1. RAD Sequencing of Four Indigenous Duck Populations
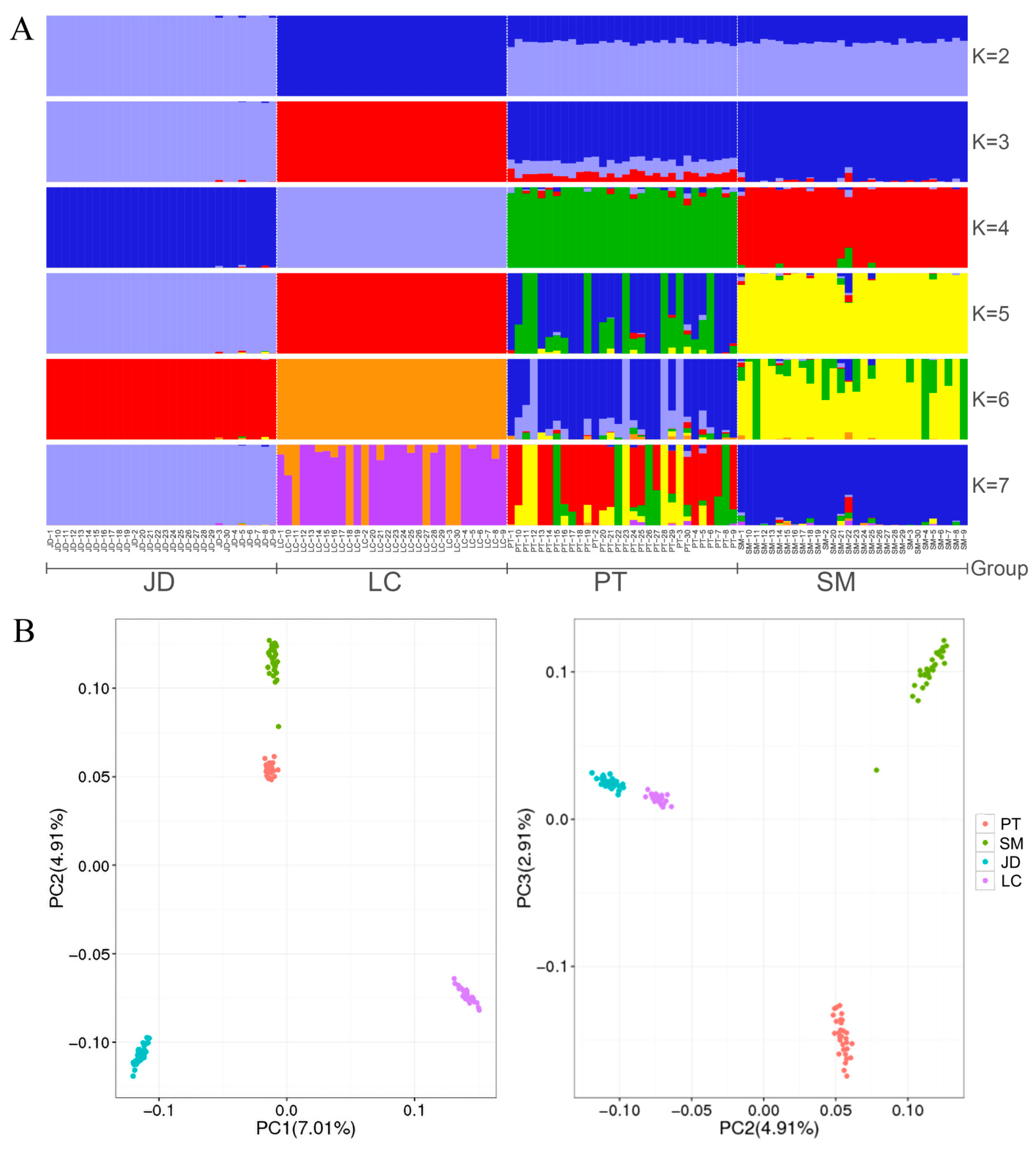
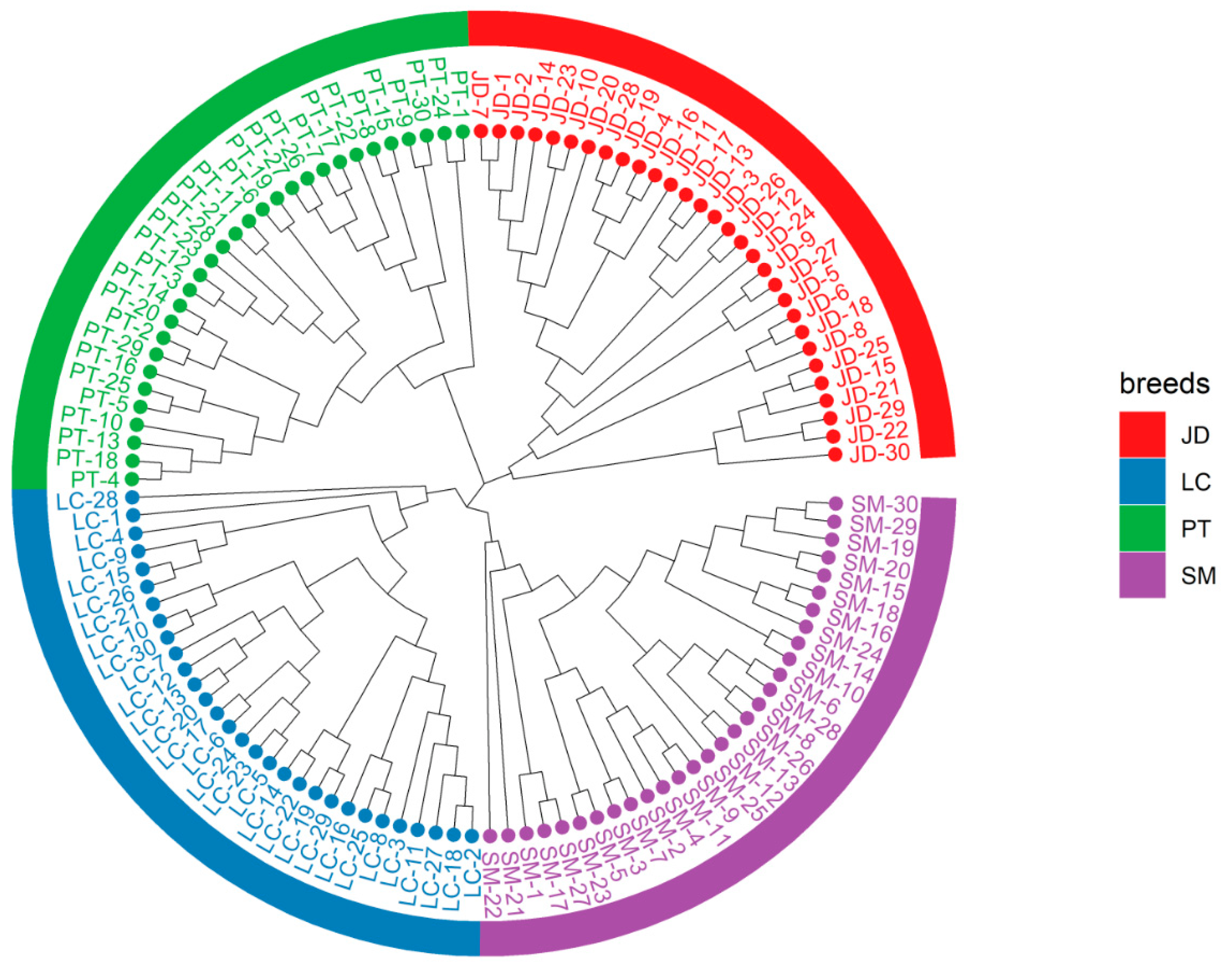
3.2. Population Genetic Structure of Four Indigenous Duck Breeds
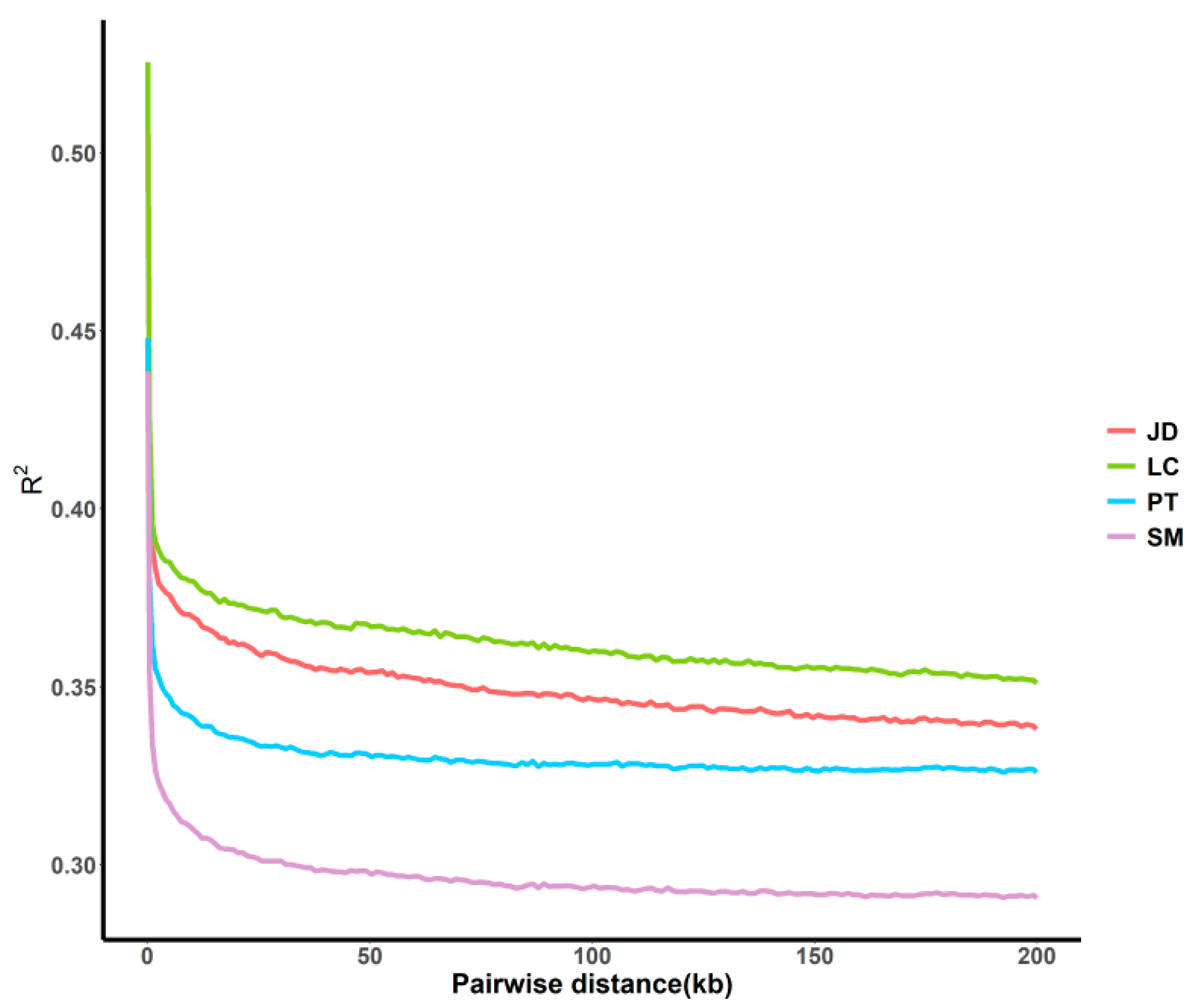
3.3. Evaluation of Genetic Diversity by Linkage Disequilibrium Trend
3.4. Runs of Homozygosity Detection and Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, R.L.; Chen, H.P.; Rouvier, R.; Poivey, J.P. Selection and Crossbreeding in Relation to Plumage Color Inheritance in Three Chinese Egg Type Duck Breeds (Anas platyrhynchos). Asian-Australas. J. Anim. Sci. 2014, 27, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Song, W.; Shu, J.; Chen, K.; Zhu, W.; Han, W.; Xu, W. Genetic diversity and population structure of 10 Chinese indigenous egg-type duck breeds assessed by microsatellite polymorphism. J. Genet. 2010, 89, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Lauvie, A.; Couix, N.; Verrier, E. No Development, No Conservation: Elements from the Conservation of Farm Animal Genetic Resources. Soc. Nat. Res. 2014, 27, 1331–1338. [Google Scholar] [CrossRef]
- Sponenberg, D.P.; Martin, A.; Couch, C.; Beranger, J. Conservation Strategies for Local Breed Biodiversity. Diversity 2019, 11, 177. [Google Scholar] [CrossRef]
- Deniskova, T.; Dotsev, A.; Lushihina, E.; Shakhin, A.; Kunz, E.; Medugorac, I.; Reyer, H.; Wimmers, K.; Khayatzadeh, N.; Soelkner, J.; et al. Population Structure and Genetic Diversity of Sheep Breeds in the Kyrgyzstan. Front. Genet. 2019, 10, 1311. [Google Scholar] [CrossRef]
- Zhang, Z.; Jia, Y.; Almeida, P.; Mank, J.E.; van Tuinen, M.; Wang, Q.; Jiang, Z.; Chen, Y.; Zhan, K.; Hou, S.; et al. Whole-genome resequencing reveals signatures of selection and timing of duck domestication. Gigascience 2018, 7, giy027. [Google Scholar] [CrossRef]
- Guo, X.; He, X.; Chen, H.; Wang, Z.; Li, H.; Wang, J.; Wang, M.; Jiang, R. Revisiting the evolutionary history of domestic and wild ducks based on genomic analyses. Zool. Res. 2021, 42, 43–50. [Google Scholar] [CrossRef]
- Zhu, T.; Qi, X.; Chen, Y.; Wang, L.; Lv, X.; Yang, W.; Zhang, J.; Li, K.; Ning, Z.; Jiang, Z.; et al. Positive selection of skeleton-related genes during duck domestication revealed by whole genome sequencing. BMC Ecol. Evol. 2021, 21, 165. [Google Scholar] [CrossRef]
- Wang, L.; Guo, J.; Xi, Y.; Ma, S.; Li, Y.; He, H.; Wang, J.; Han, C.; Bai, L.; Mustafa, A.; et al. Understanding the Genetic Domestication History of the Jianchang Duck by Genotyping and Sequencing of Genomic Genes under Selection. G3 2020, 10, 1469–1476. [Google Scholar] [CrossRef]
- Wang, R.; Sun, J.; Han, H.; Huang, Y.; Chen, T.; Yang, M.; Wei, Q.; Wan, H.; Liao, Y. Whole-genome resequencing reveals genetic characteristics of different duck breeds from the Guangxi region in China. G3 2021, 11, jkab054. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Molyte, A.; Urnikyte, A.; Kucinskas, V. A comparative analysis of mathematical methods for homogeneity estimation of the Lithuanian population. Acta Med. Litu. 2019, 26, 211–216. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef]
- Lavretsky, P.; McInerney, N.R.; Mohl, J.E.; Brown, J.I.; James, H.F.; McCracken, K.G.; Fleischer, R.C. Assessing changes in genomic divergence following a century of human-mediated secondary contact among wild and captive-bred ducks. Mol. Ecol. 2020, 29, 578–595. [Google Scholar] [CrossRef]
- Nothnagel, M.; Lu, T.T.; Kayser, M.; Krawczak, M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum. Mol. Genet. 2010, 19, 2927–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, J.; Seguel, M.; Saenz-Agudelo, P.; Acosta-Jamett, G.; Verdugo, C. Genetic diversity and kinship relationships in one of the largest South American fur seal (Arctocephalus australis) populations of the Pacific Ocean. Ecol. Evol. 2021, 11, 8743–8753. [Google Scholar] [CrossRef]
- Su, Y.; Chen, G. DNA microsatellite analysis of genetic diversity among Chinese indigenous laying-type ducks (Anas platyrhynchos). Czech J. Anim. Sci. 2009, 54, 128–135. [Google Scholar] [CrossRef]
- Li, H.; Zhu, W.; Song, W.; Shu, J.; Han, W.; Chen, K. Origin and genetic diversity of Chinese domestic ducks. Mol. Phylogenet. Evol. 2010, 57, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Y.; Jiang, X.; Zhong, Z.; Xu, Q.; Chen, G. Population Genetic Structure Analysis of Indigenous Ducks in Fujian Province. China Poult. 2013, 35, 11–15. (In Chinese) [Google Scholar]
- Xu, L.; Zhu, B.; Wang, Z.; Xu, L.; Liu, Y.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Zhang, S.; et al. Evaluation of Linkage Disequilibrium, Effective Population Size and Haplotype Block Structure in Chinese Cattle. Animals 2019, 9, 83. [Google Scholar] [CrossRef]
- Gorssen, W.; Meyermans, R.; Janssens, S.; Buys, N. A publicly available repository of ROH islands reveals signatures of selection in different livestock and pet species. Genet. Sel. Evol. 2021, 53, 2. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, C.; Crooijmans, R.P.M.A.; Bosse, M.; Hiemstra, S.J.; Groenen, M.A.M.; Megens, H.-J. The effects of recent changes in breeding preferences on maintaining traditional Dutch chicken genomic diversity. Heredity 2018, 121, 564–578. [Google Scholar] [CrossRef]
- Marchesi, J.A.P.; Buzanskas, M.E.; Cantao, M.E.; Ibelli, A.M.G.; Peixoto, J.O.; Joaquim, L.B.; Moreira, G.C.M.; Godoy, T.F.; Sbardella, A.P.; Figueiredo, E.A.P.; et al. Relationship of runs of homozygosity with adaptive and production traits in a paternal broiler line. Animal 2018, 12, 1126–1134. [Google Scholar] [CrossRef]
- Wang, M.-S.; Li, Y.; Peng, M.-S.; Zhong, L.; Wang, Z.-J.; Li, Q.-Y.; Tu, X.-L.; Dong, Y.; Zhu, C.-L.; Wang, L.; et al. Genomic Analyses Reveal Potential Independent Adaptation to High Altitude in Tibetan Chickens. Mol. Biol. Evol. 2015, 32, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic Patterns of Homozygosity in Worldwide Human Populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef]
- Ablondi, M.; Dadousis, C.; Vasini, M.; Eriksson, S.; Mikko, S.; Sabbioni, A. Genetic Diversity and Signatures of Selection in a Native Italian Horse Breed Based on SNP Data. Animals 2020, 10, 1005. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Sun, H.; Zhang, Z.; Zhao, Q.; Olasege, B.S.; Li, Q.; Yue, Y.; Ma, P.; Zhang, X.; Wang, Q.; et al. Assessment of Autozygosity Derived from Runs of Homozygosity in Jinhua Pigs Disclosed by Sequencing Data. Front. Genet. 2019, 10, 274. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, W.; Li, M.; Gao, P.; Hei, W.; He, Z.; Wu, Y.; Liu, J.; Cai, C.; Li, B.; et al. Transcriptome profile of skeletal muscle at different developmental stages in Large White and Mashen pigs. Can. J. Anim. Sci. 2019, 99, 867–880. [Google Scholar] [CrossRef]
- Oczkowicz, M.; Ropka-Molik, K.; Tyra, M. Analysis of the associations between polymorphisms in GNAS complex locus and growth, carcass and meat quality traits in pigs. Mol. Biol. Rep. 2013, 40, 6419–6427. [Google Scholar] [CrossRef]
- Imumorin, I.G.; Kim, E.-H.; Lee, Y.-M.; De Koning, D.-J.; van Arendonk, J.; De Donato, M.; Taylor, J.F.; Kim, J.-J. Genome scan for parent-of-origin QTL effects on bovine growth and carcass traits. Front. Genet. 2011, 2, 44. [Google Scholar] [CrossRef]
- Liu, H.; Luo, Q.; Zhang, J.; Mo, C.; Wang, Y.; Li, J. Endothelins (EDN1, EDN2, EDN3) and their receptors (EDNRA, EDNRB, EDNRB2) in chickens: Functional analysis and tissue distribution. Gen. Comp. Endocrinol. 2019, 283, 113231. [Google Scholar] [CrossRef]
- Chen, X.; Bai, X.; Liu, H.; Zhao, B.; Yan, Z.; Hou, Y.; Chu, Q. Population Genomic Sequencing Delineates Global Landscape of Copy Number Variations that Drive Domestication and Breed Formation of in Chicken. Front. Genet. 2022, 13, 830393. [Google Scholar] [CrossRef]
- Li, L.; Li, D.; Liu, L.; Li, S.; Feng, Y.; Peng, X.; Gong, Y. Endothelin Receptor B2 (EDNRB2) Gene Is Associated with Spot Plumage Pattern in Domestic Ducks (Anas platyrhynchos). PLoS ONE 2015, 10, e0125883. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Li, J.; Zhao, F.; Zhou, M.; Wang, J.; Xiao, T. Transcriptome analysis of genes potentially associated with white and black plumage formation in Chinese indigenous ducks (Anas platyrhynchos). Br. Poult. Sci. 2022, 63, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, T.; Zhu, M.; Wang, R.; Huang, Y.; Wei, Q.; Yang, M.; Liao, Y. Whole-genome sequencing revealed genetic diversity and selection of Guangxi indigenous chickens. PLoS ONE 2022, 17, e0250392. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Li, J.; Zhang, Y.; Han, D.; Hua, G.; Wang, J.; Deng, X.; Wu, C. Genomic Analysis Reveals Pleiotropic Alleles at EDN3 and BMP7 Involved in Chicken Comb Color and Egg Production. Front. Genet. 2019, 10, 612. [Google Scholar] [CrossRef]
- Hou, H.; Wang, X.; Zhang, C.; Tu, Y.; Lv, W.; Cai, X.; Xu, Z.; Yao, J.; Yang, C. Genomic analysis of GBS data reveals genes associated with facial pigmentation in Xinyang blue-shelled layers. Arch. Anim. Breed. 2020, 63, 483–491. [Google Scholar] [CrossRef]
- Beaubien, F.; Raja, R.; Kennedy, T.E.; Fournier, A.E.; Cloutier, J.-F. Slitrk1 is localized to excitatory synapses and promotes their development. Sci. Rep. 2016, 6, 27343. [Google Scholar] [CrossRef]
- Tekin, M.; Chioza, B.A.; Matsumoto, Y.; Diaz-Horta, O.; Cross, H.E.; Duman, D.; Kokotas, H.; Moore-Barton, H.L.; Sakoori, K.; Ota, M.; et al. SLITRK6 mutations cause myopia and deafness in humans and mice. J. Clin. Investig. 2013, 123, 2094–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Num of SNPs | Start | End | Length (bp) | Genes |
|---|---|---|---|---|---|
| 1 | 108 | 45626183 | 50131802 | 4505620 | LOC113839685, LOC106014954, LOC113839687, LOC113839688, LOC113839689, SLITRK1, LOC113839691, LOC113844965, LOC113839693, SLITRK6, LOC106017061, LOC113839694, SLITRK5, LOC110354298, MRPS12, LOC106019638, LOC110352518 |
| 1 | 51 | 70797389 | 72148337 | 1350949 | LOC101791573, LOC113840661, LOC106016827, LOC106016828, NLGN4X |
| 1 | 66 | 97150638 | 98972361 | 1821724 | GABPA, ATP5PF, JAM2, MRPL39, LOC106014525, LOC106014508, LOC106014510, LOC110351253, LOC106014431, LOC113842349, NCAM2 |
| 3 | 66 | 88621432 | 89675961 | 1054530 | LOC106016669 |
| 21 | 50 | 10214904 | 11278269 | 1063366 | LOC113845753, LOC110353825, LOC106019012, C21H20orf85, ANKRD60, LOC101805171, RAB22A, VAPB, APCDD1L, STX16, NPEPL1, LOC113839645, LOC106018983, GNAS, LOC101792091, LOC106018981, LOC113839646, NELFCD, CTSZ, TUBB1, ATP5F1E, LOC106018967, PRELID3B, ZNF831, EDN3, PHACTR3, SYCP2, LOC101790963, FAM217B, LOC113839657 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, R.; Li, J.; Yang, Y.; Yang, Y.; Chen, J.; Zhao, F.; Xiao, T. Genome-Wide Population Structure Analysis and Genetic Diversity Detection of Four Chinese Indigenous Duck Breeds from Fujian Province. Animals 2022, 12, 2302. https://doi.org/10.3390/ani12172302
Lin R, Li J, Yang Y, Yang Y, Chen J, Zhao F, Xiao T. Genome-Wide Population Structure Analysis and Genetic Diversity Detection of Four Chinese Indigenous Duck Breeds from Fujian Province. Animals. 2022; 12(17):2302. https://doi.org/10.3390/ani12172302
Chicago/Turabian StyleLin, Ruiyi, Jiaquan Li, Yue Yang, Yinhua Yang, Jimin Chen, Fanglu Zhao, and Tianfang Xiao. 2022. "Genome-Wide Population Structure Analysis and Genetic Diversity Detection of Four Chinese Indigenous Duck Breeds from Fujian Province" Animals 12, no. 17: 2302. https://doi.org/10.3390/ani12172302
APA StyleLin, R., Li, J., Yang, Y., Yang, Y., Chen, J., Zhao, F., & Xiao, T. (2022). Genome-Wide Population Structure Analysis and Genetic Diversity Detection of Four Chinese Indigenous Duck Breeds from Fujian Province. Animals, 12(17), 2302. https://doi.org/10.3390/ani12172302