
Metagenomic Comparisons between Soft and Hard Feces of Plateau Pikas (Ochotona curzoniae)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Processing and 16S rDNA Amplicon Sequencing
2.3. Bioinformatics and Statistical Analysis
3. Results
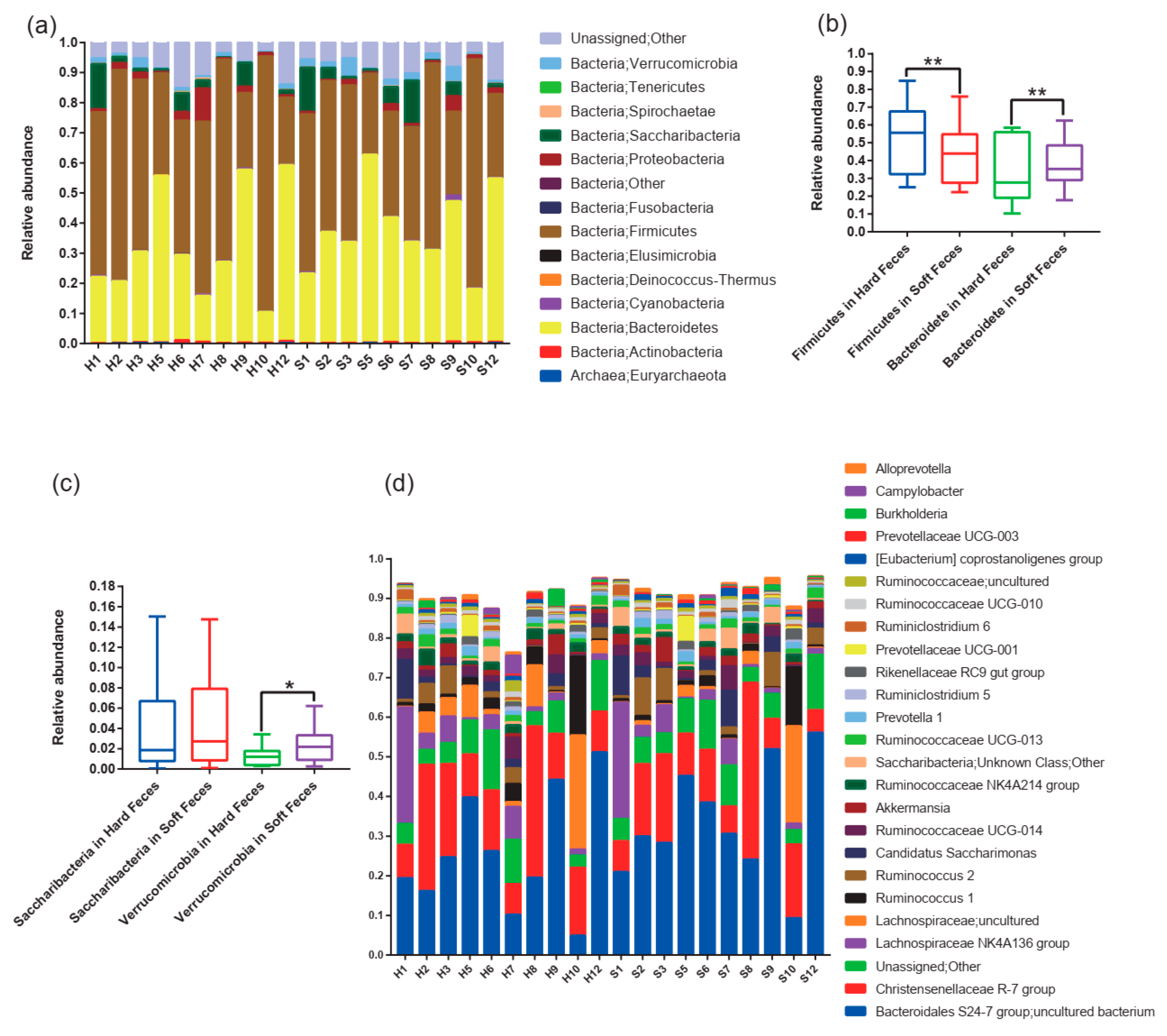
3.1. Gut Microbiota Composition of Plateau Pikas
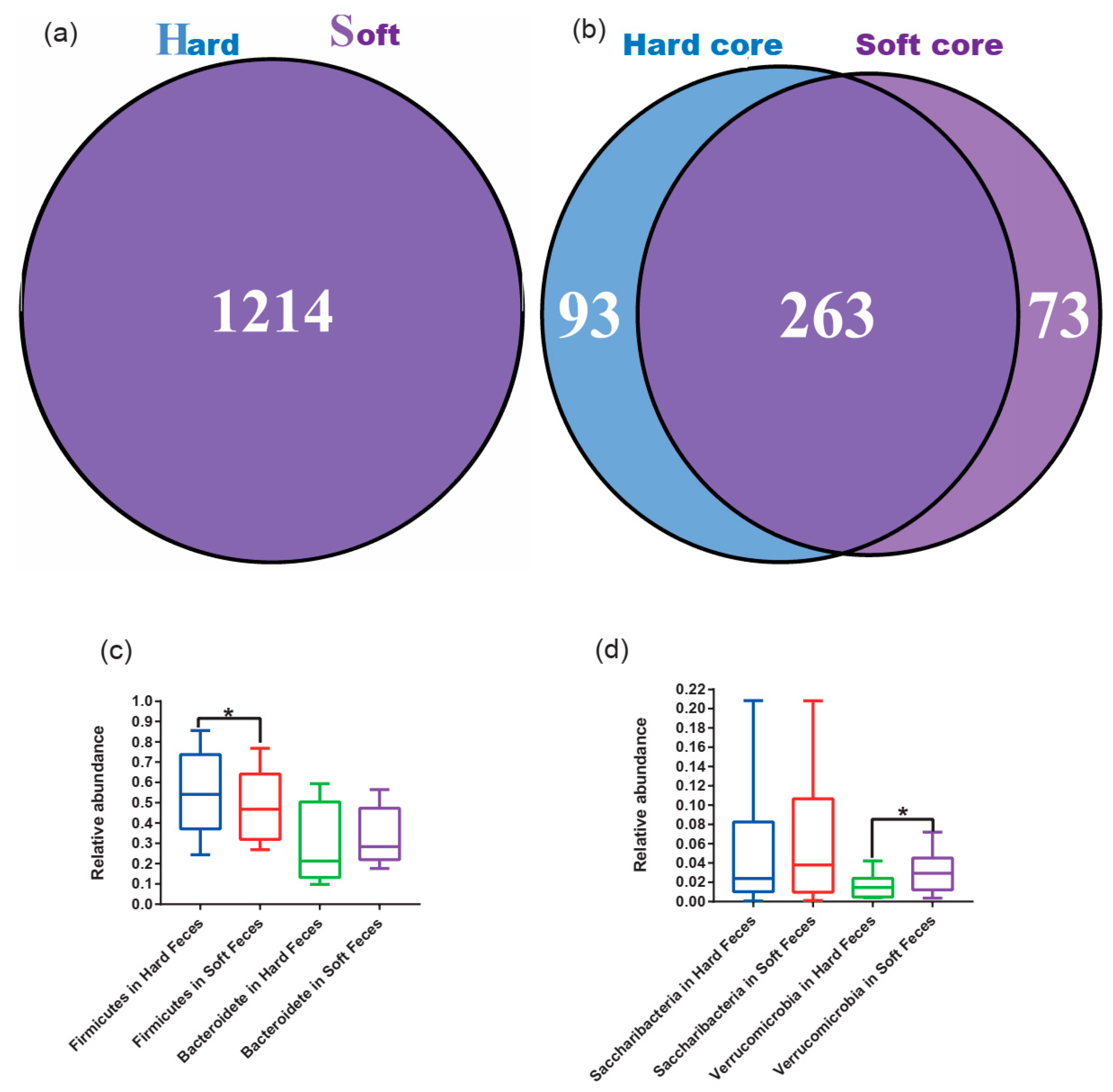
3.2. Microbial Diversity of Pika
3.3. Core Microbial Community Better Reveals the Discrepancy between Hard and Soft Feces
3.4. The Predicted Function of Gut Microbial Communities in Hard and Soft Feces
3.5. KEGG Pathway Annotation and Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, X.; Zhao, L.; Xu, T.; Xu, S. The plateau pika has multiple benefits for alpine grassland ecosystem in Qinghai–Tibet Plateau. Ecosyst. Health Sustain. 2020, 6, 1750973. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.T.; Badingqiuying; Wilson, M.C.; Hogan, B.W. Functional-trait ecology of the plateau pika Ochotona curzoniae in the Qinghai-Tibetan Plateau ecosystem. Integr. Zool. 2019, 14, 87–103. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.H.; Smith, A.T. Keystone status of plateau pikas (Ochotona curzoniae): Effect of control on biodiversity of native birds. Biodivers. Conserv. 2003, 12, 1901–1912. [Google Scholar] [CrossRef]
- Dahal, N.; Lissovsky, A.A.; Lin, Z.; Solari, K.; Hadly, E.A.; Zhan, X.; Ramakrishnan, U. Genetics, morphology and ecology reveal a cryptic pika lineage in the Sikkim Himalaya. Mol. Phylogenet. Evol. 2017, 106, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Zheng, C.L.; Zhang, Y.P.; Li, W.H. Molecular systematics of pikas (genus Ochotona) inferred from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2000, 16, 85–95. [Google Scholar] [CrossRef]
- Wang, J.M.; Zhang, Y.M.; Wang, D.H. Seasonal regulations of energetics, serum concentrations of leptin, and uncoupling protein 1 content of brown adipose tissue in root voles (Microtus oeconomus) from the Qinghai-Tibetan plateau. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 2006, 176, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Zhang, Y.M.; Wang, D.H. Seasonal thermogenesis and body mass regulation in plateau pikas (Ochotona curzoniae). Oecologia 2006, 149, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.P.; Li, W.J.; Yang, M.; Ji, W.H.; Zhang, Y.M. Life history of the plateau pika (Ochotona curzoniae) in alpine meadows of the Tibetan Plateau. Mamm. Biol. 2013, 78, 68–72. [Google Scholar] [CrossRef]
- Clauss, M.; Steuer, P.; Muller, D.W.H.; Codron, D.; Hummel, J. Herbivory and Body Size: Allometries of Diet Quality and Gastrointestinal Physiology, and Implications for Herbivore Ecology and Dinosaur Gigantism. PLoS ONE 2013, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, H. Coprophagy in leporids and other mammalian herbivores. Mammal Rev. 2001, 31, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Pickard, D.W.; Stevens, C.E. Digesta flow through rabbit large-intestine. Am. J. Physiol. 1972, 222, 1161–1166. [Google Scholar] [CrossRef]
- Naumova, E.I.; Zharova, G.K.; Chistova, T.Y.; Formozov, N.A. The structure of the epithelial surface of the gastrointestinal tract of Pikas (Ochotona pallasi and O-dauurica, Lagomorpha, Ochotonidae): Functional and species specificity. Biol. Bull. 2014, 41, 357–363. [Google Scholar] [CrossRef]
- Pshennikov, A.E.; Alekseev, V.G.; Koryakin, I.I.; Gnutov, D.Y. Coprophagy in ochotona-hyperboreae in yakutiya. Zool. Zhurnal 1990, 69, 106–114. [Google Scholar]
- Björnhag, G.; Snipes, R.L. Colonic Separation Mechanism in Lagomorph and Rodent Species—A Comparison. Zoosyst. Evol. 1999, 75, 275–281. [Google Scholar] [CrossRef]
- Zeng, B.; Han, S.S.; Wang, P.; Wen, B.; Jian, W.S.; Guo, W.; Yu, Z.J.; Du, D.; Fu, X.C.; Kong, F.L.; et al. The bacterial communities associated with fecal types and body weight of rex rabbits. Sci. Rep. 2015, 5, 8. [Google Scholar] [CrossRef]
- Bjornhag, G. The retrograde transport of fluid in the proximal colon of rabbits. Swed. J. Agric. Res. 1981, 11, 63–69. [Google Scholar]
- Ruckebusch, Y.; Hornicke, H. Motility of rabbits colon and cecotrophy. Physiol. Behav. 1977, 18, 871–878. [Google Scholar] [CrossRef]
- Ehrlein, H.J.; Reich, H.; Schwinger, M. Colonic motility and transit of digesta during hard and soft feces formation in rabbits. J. Physiol. 1983, 338, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Eden, A. Coprophagy in the rabbit—Origin of ‘night’ faeces. Nature 1940, 145, 628–629. [Google Scholar] [CrossRef]
- Southern, H.N. Coprophagy in the wild rabbit. Nature 1940, 145, 262. [Google Scholar] [CrossRef]
- Bjornhag, G. Adaptations in the Large Intestine Allowing Small Animals to Eat Fibrous Foods; Cambridge University Press: Cambridge, UK, 1994; pp. 287–309. [Google Scholar]
- Eden, A. Coprophagy in the rabbit. Nature 1940, 145, 36–37. [Google Scholar] [CrossRef]
- Barnes, R.H. Nutritional implications of coprophagy. Nutr. Rev. 1962, 20, 289–291. [Google Scholar] [CrossRef]
- Sharkey, M.J. Some aspects of coprophagy in rabbits and guinea pigs fed fresh lucerne. Mammalia 1971, 35, 162–168. [Google Scholar] [CrossRef]
- Kizilova, A.K.; Kravchenko, I.K. Diversity of diazotrophic gut inhabitants of pikas (Ochotonidae) revealed by PCR-DGGE analysis. Microbiology 2014, 83, 288–295. [Google Scholar] [CrossRef]
- Bo, T.B.; Zhang, X.Y.; Kohl, K.D.; Wen, J.; Tian, S.J.; Wang, D.H. Coprophagy prevention alters microbiome, metabolism, neurochemistry, and cognitive behavior in a small mammal. ISME J. 2020, 14, 2625–2645. [Google Scholar] [CrossRef]
- Chevalier, C.; Stojanovic, O.; Colin, D.J.; Suarez-Zamorano, N.; Tarallo, V.; Veyrat-Durebex, C.; Rigo, D.; Fabbiano, S.; Stevanovic, A.; Hagemann, S.; et al. Gut Microbiota Orchestrates Energy Homeostasis during Cold. Cell 2015, 163, 1360–1374. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Zhang, L.; Fan, C.; Li, W.; Liu, C.; Zhang, H.; Cheng, Q.; Zhang, Y. Sympatric Yaks and Plateau Pikas Promote Microbial Diversity and Similarity by the Mutual Utilization of Gut Microbiota. Microorganisms 2021, 9, 1890. [Google Scholar] [CrossRef]
- Fu, H.; Zhang, L.; Fan, C.; Liu, C.; Li, W.; Cheng, Q.; Zhao, X.; Jia, S.; Zhang, Y. Environment and host species identity shape gut microbiota diversity in sympatric herbivorous mammals. Microb. Biotechnol. 2020, 14, 1300–1315. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Moeller, A.H.; Peeters, M.; Ndjango, J.B.; Li, Y.; Hahn, B.H.; Ochman, H. Sympatric chimpanzees and gorillas harbor convergent gut microbial communities. Genome Res. 2013, 23, 1715–1720. [Google Scholar] [CrossRef] [Green Version]
- Amato, K.R.; Sanders, J.G.; Song, S.J.; Nute, M.; Metcalf, J.L.; Thompson, L.R.; Morton, J.T.; Amir, A.; McKenzie, V.J.; Humphrey, G.; et al. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 2018, 13, 12. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; Collado, M.C.; Ben-Amor, K.; Salminen, S.; de Vos, W.M. The mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl. Environ. Microbiol. 2008, 74, 1646–1648. [Google Scholar] [CrossRef] [Green Version]
- Georgiades, K.; Merhej, V.; Raoult, D. The influence or rickettsiologists on post-modern microbiology. Front. Cell. Infect. Microbiol. 2011, 1, 12. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, H. Coprophagy in the japanese hare (lepus-brachyurus)—Reingestion of all the hard and soft feces during the daytime stay in the form. J. Zool. 1994, 232, 447–456. [Google Scholar] [CrossRef]
- Mervyn, G.; David, D. The role of the soft pellets in the production of lactic acid in the rabbit stomach. J. Nutr. 1963, 80, 171–180. [Google Scholar]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.F.; Wu, Q.; Dai, J.Y.; Zhang, S.N.; Wei, F.W. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 17714–17719. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Mota, R.; Kohl, K.D.; Orr, T.J.; Denise Dearing, M. Natural diets promote retention of the native gut microbiota in captive rodents. ISME J. 2020, 14, 67–78. [Google Scholar] [CrossRef]
- Hirakawa, H. The Formation and Passage of Soft and Hard Feces in the Hindgut of the Japanese Hare Lepus brachyurus. J. Mammal. Soc. Jpn. 1995, 20, 89–94. [Google Scholar]
- Pshennikov, A.E.; Borisov, Z.Z.; Vasiliev, I.S. Coprophagy and its rhythms in mountain hare (lepus-timidus) of central Yakutia. Zool. Zhurnal 1988, 67, 1357–1362. [Google Scholar]
- Formozov, N.A.; Kizilova, A.K.; Panteleeva, A.N.; Naumova, E.I. Nitrogen fixation as a possible physiological basis of coprophagy in pikas (Ochotona, Lagomorpha, Mammalia). Dokl. Biol. Sci. 2012, 443, 126–129. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, H.; Li, W. Metagenomic Comparisons between Soft and Hard Feces of Plateau Pikas (Ochotona curzoniae). Animals 2022, 12, 149. https://doi.org/10.3390/ani12020149
Fu H, Li W. Metagenomic Comparisons between Soft and Hard Feces of Plateau Pikas (Ochotona curzoniae). Animals. 2022; 12(2):149. https://doi.org/10.3390/ani12020149
Chicago/Turabian StyleFu, Haibo, and Wenjing Li. 2022. "Metagenomic Comparisons between Soft and Hard Feces of Plateau Pikas (Ochotona curzoniae)" Animals 12, no. 2: 149. https://doi.org/10.3390/ani12020149