
Integrated Transcriptome Analysis Reveals the Crucial mRNAs and miRNAs Related to Fecundity in the Hypothalamus of Yunshang Black Goats during the Luteal Phase


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
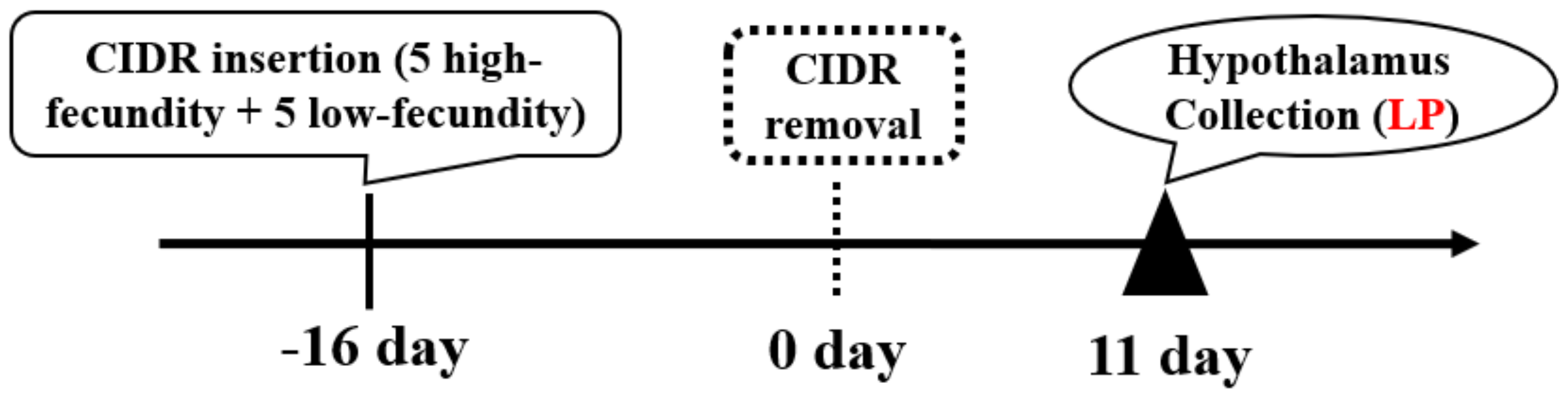
2.2. Goat Selection and Sample Collection
2.3. Total RNA Extraction
2.4. Library Preparation and Sequencing
2.5. Analysis of the Differential Expression mRNAs and miRNAs
2.6. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Analyses
2.7. Integrated miRNA-mRNA Co-Expression Network Analysis
2.8. RT-qPCR Validation
2.9. Statistical Analysis
3. Results
3.1. The cDNA Library Sequencing and mRNA Transcriptome Analysis
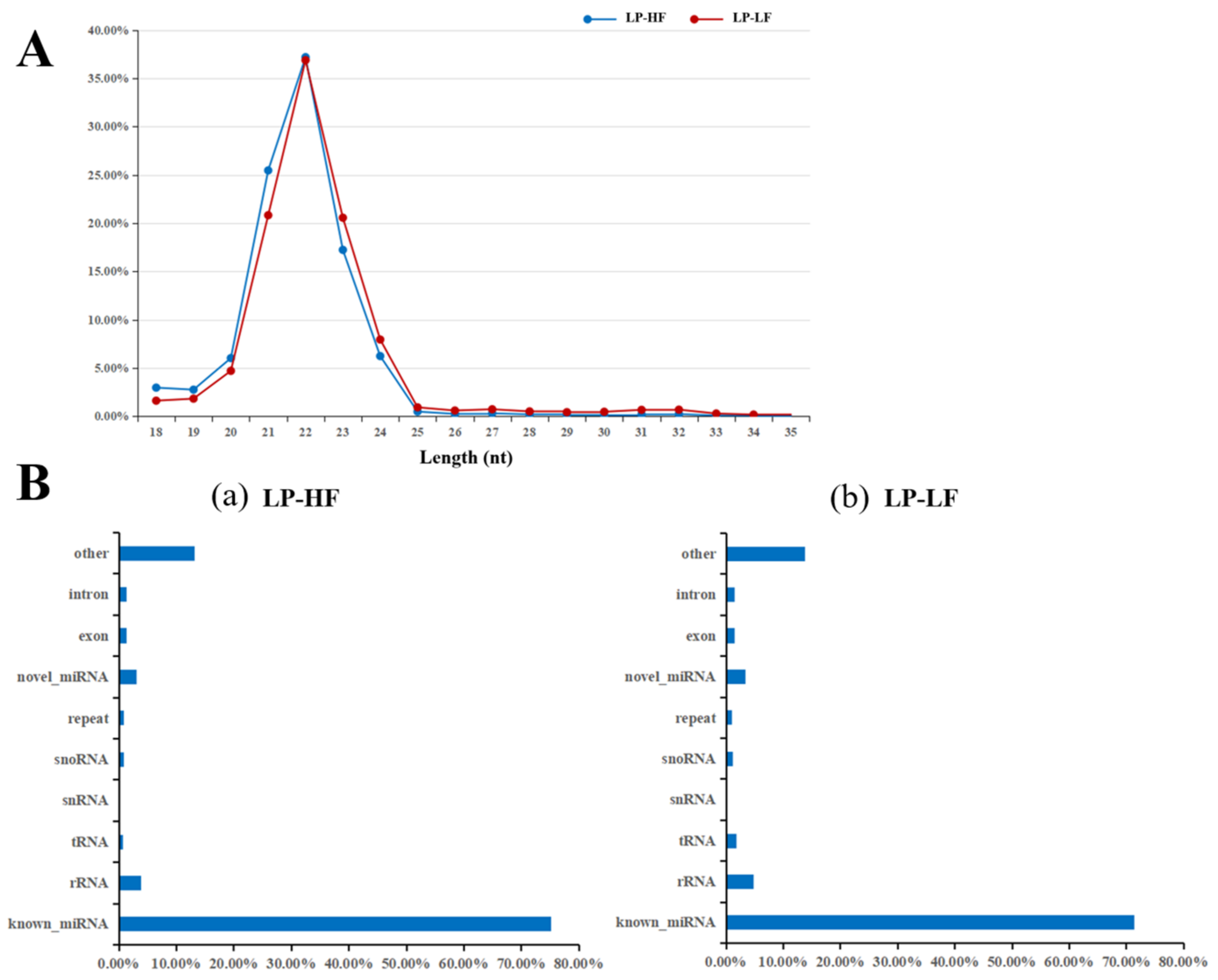
3.2. Small RNA Library Sequencing and miRNA Transcriptome Analysis
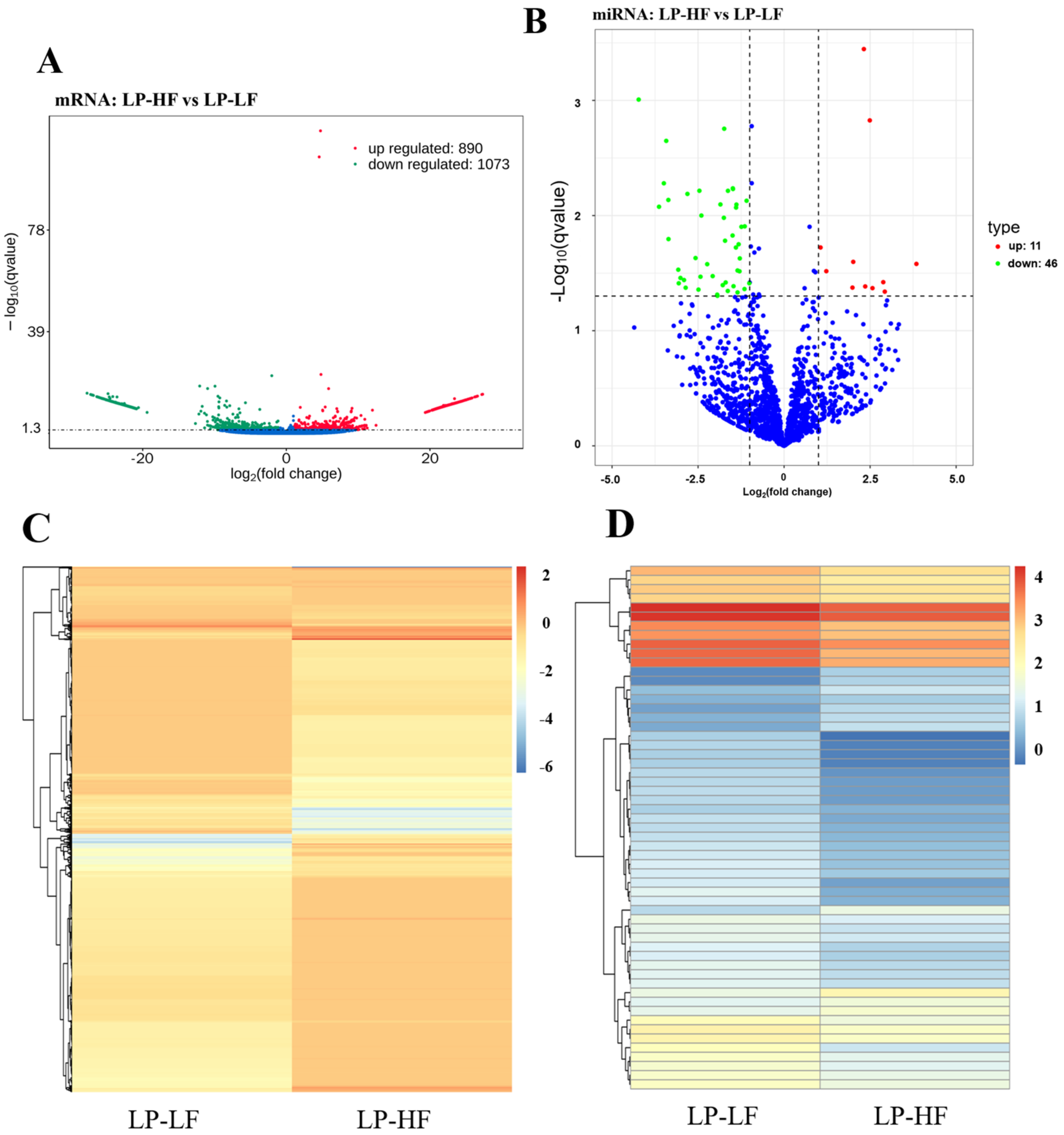
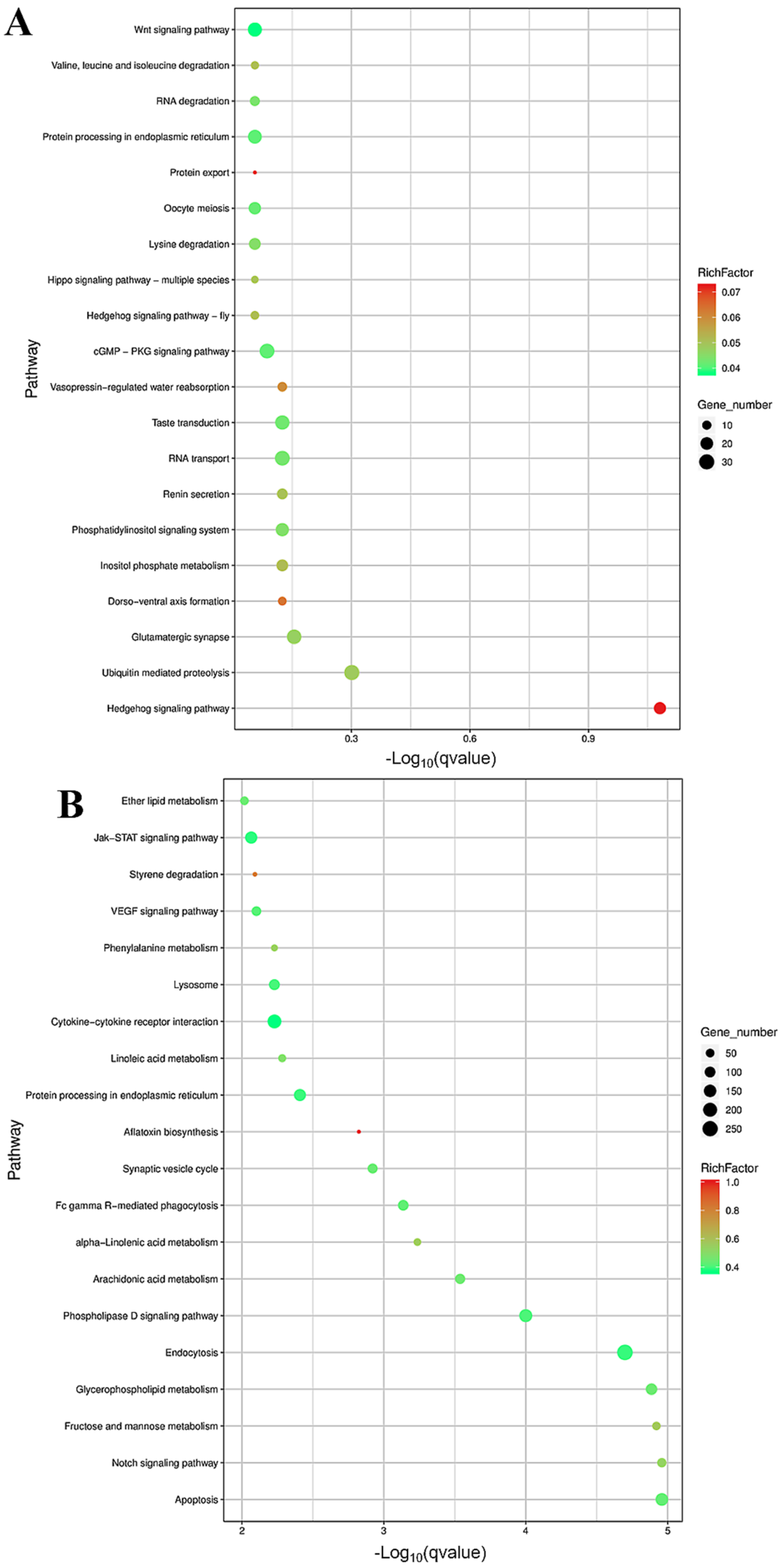
3.3. Differential Expression and Functional Enrichment Analysis of the mRNAs in the LP-HF vs. LP-LF
3.4. Differential Expression and Functional Enrichment Analysis of the miRNAs in the LP-HF vs. LP-LF
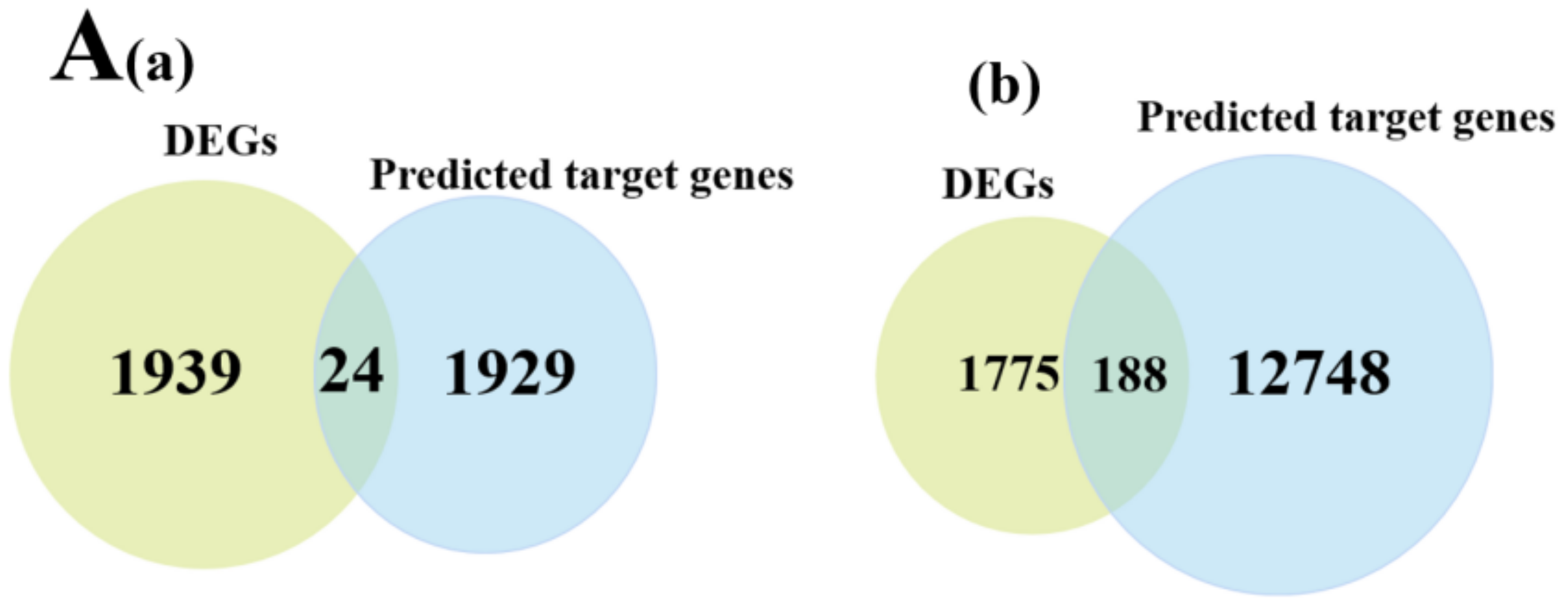
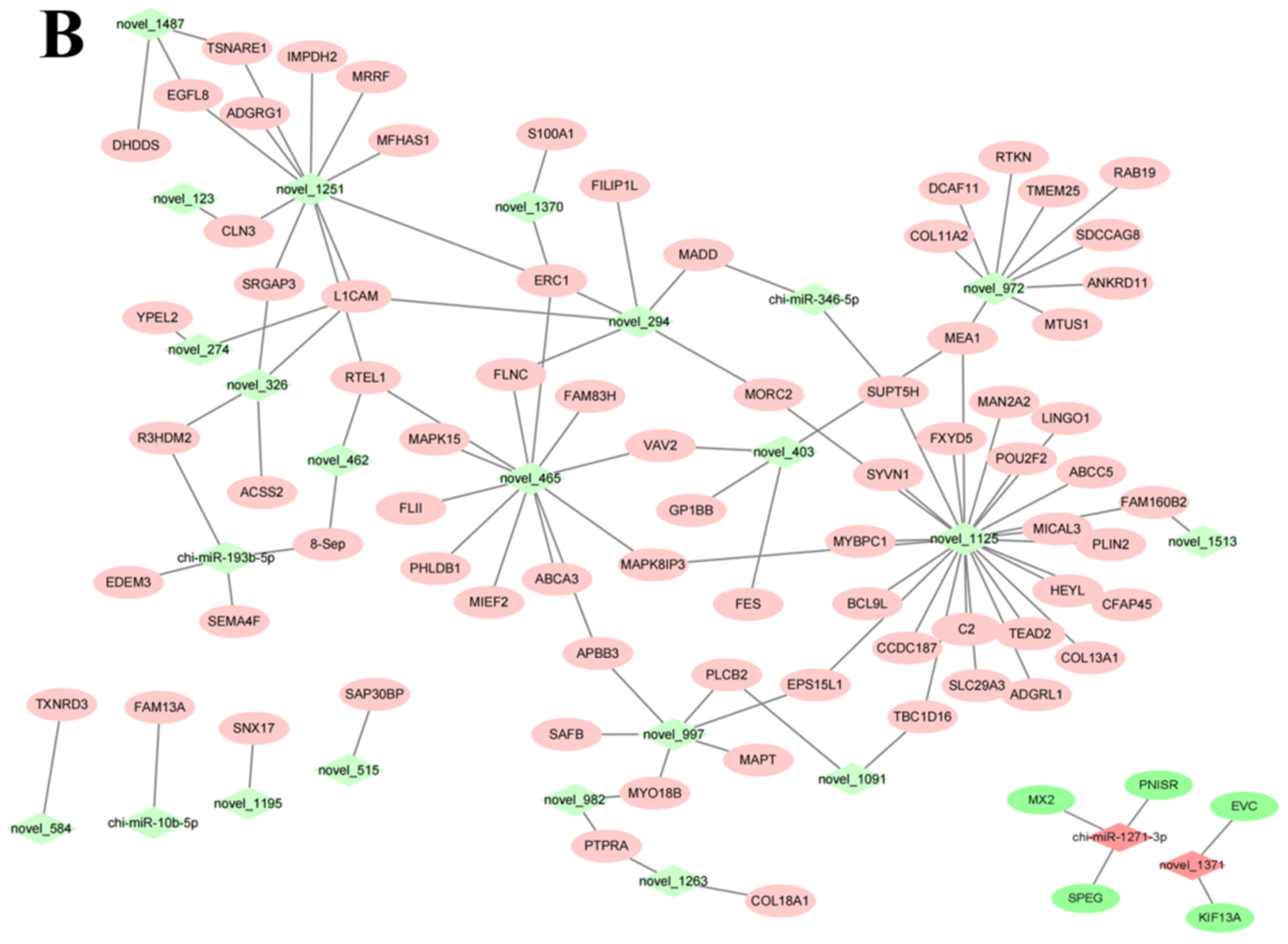
3.5. miRNA-mRNA Co-Expression Network Analysis
3.6. Data Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen-Tannoudji, J.; Avet, C.; Garrel, G.; Counis, R.; Simon, V. Decoding high Gonadotropin-releasing hormone pulsatility: A role for GnRH receptor coupling to the cAMP pathway? Front. Endocrinol. 2012, 3, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larder, R.; Kimura, I.; Meadows, J.; Clark, D.D.; Mayo, S.; Mellon, P.L. Gene dosage of Otx2 is important for fertility in male mice. Mol. Cell. Endocrinol. 2013, 377, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.; Ding, Y.; Kong, X.; Feng, G.; Xiang, W.; Chen, L.; Yang, F.; Zhang, K.; Chu, M.; Wang, P.; et al. Reproductive role of miRNA in the hypothalamic-pituitary axis. Mol. Cell. Neurosci. 2018, 88, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, W. Hypothalamic and pituitary transcriptome profiling using RNA-sequencing in high-yielding and low-yielding laying hens. Sci. Rep. 2019, 9, 10285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Sun, M.; Shan, Y.; Zheng, X.; Ma, H.; Ma, W.; Wang, Z.; Pei, X.; Wang, Y. Endoplasmic Reticulum Stress-Mediated Apoptotic Pathway Is Involved in Corpus Luteum Regression in Rats. Reprod. Sci. 2015, 22, 572–584. [Google Scholar] [CrossRef] [Green Version]
- Devoto, L.; Henríquez, S.; Kohen, P.; Strauss, J.F., 3rd. The significance of estradiol metabolites in human corpus luteum physiology. Steroids 2017, 123, 50–54. [Google Scholar] [CrossRef]
- Samaan, S.; Tranchevent, L.-C.; Dardenne, E.; Espinoza, M.P.; Zonta, E.; Germann, S.; Gratadou, L.; Dutertre, M.; Auboeuf, D. The Ddx5 and Ddx17 RNA helicases are cornerstones in the complex regulatory array of steroid hormone-signaling pathways. Nucleic Acids Res. 2013, 42, 2197–2207. [Google Scholar] [CrossRef]
- Piper, L.R.; Bindon, B.M. The Booroola Merino and the performance of medium non-Peppin crosses at Armidale. Wool Technol. Sheep Breed. 1983, 31, 14–19. [Google Scholar]
- Huang, D.W.; Di, R.; Hu, W.P.; Wang, X.Y.; Pan, Z.Y.; Guo, X.F.; Cao, X.H.; Liu, Q.Y.; Chu, M.X. Expression analysis of DIO2 and DIO3 genes related to repro-ductive seasonality in goats (Capra hircus). Chin. J. Agric. Biotechol. 2016, 24, 1536–1543. [Google Scholar]
- Pinilla, L.; Aguilar, E.; Dieguez, C.; Millar, R.P.; Tena-Sempere, M. Kisspeptins and Reproduction: Physiological Roles and Regulatory Mechanisms. Physiol. Rev. 2012, 92, 1235–1316. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, V.M. Metabolic regulation of kisspeptin—The link between energy balance and reproduction. Nat. Rev. Endocrinol. 2020, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Herzer, S.; Silahtaroglu, A.; Meister, B. Locked nucleic acid-based in situ hybridisation reveals miR-7a as a hypothala-mus-enriched microRNA with a distinct expression pattern. J. Neuroendocrinol. 2012, 24, 1492–1504. [Google Scholar] [CrossRef]
- Fang, Y.; Xue, J.-L.; Shen, Q.; Chen, J.; Tian, L. MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology 2012, 55, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; LaPierre, M.P.; Gasser, E.; Denzler, R.; Yang, Y.; Rülicke, T.; Kero, J.; Latreille, M.; Stoffel, M. Loss of microRNA-7a2 induces hypogonadotropic hy-pogonadism and infertility. J. Clin. Investig. 2017, 127, 1061–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corre, C.; Shinoda, G.; Zhu, H.; Cousminer, D.L.; Crossman, C.; Bellissimo, C.; Goldenberg, A.; Daley, G.Q.; Palmert, M.R. Sex-specific regulation of weight and puberty by the Lin28/let-7 axis. J. Endocrinol. 2016, 228, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Sangiao-Alvarellos, S.; Manfredi-Lozano, M.; Ruiz-Pino, F.; Navarro, V.M.; Sánchez-Garrido, M.A.; Leon, S.; Dieguez, C.; Cordido, F.; Matagne, V.; Dissen, G.A.; et al. Changes in hypothalamic expression of the Lin28/let-7 system and related microRNAs during postnatal maturation and after experimental manipulations of puberty. Endocrinology 2013, 154, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Heras, V.; Sangiao-Alvarellos, S.; Manfredi-Lozano, M.; Sanchez-Tapia, M.J.; Ruiz-Pino, F.; Roa, J.; Lara-Chica, M.; Morrugares-Carmona, R.; Jouy, N.; Abreu, A.P.; et al. Hypothalamic miR-30 reg-ulates puberty onset via repression of the puberty-suppressing factor, Mkrn3. PLoS Biol. 2019, 17, e3000532. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tang, J.; Di, R.; Liu, Q.; Wang, X.; Gan, S.; Zhang, X.; Zhang, J.; Chu, M.; Hu, W. Integrated hypothalamic transcriptome profiling reveals the re-productive roles of mRNAs and miRNAs in sheep. Front. Genet. 2020, 10, 1296. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yuxin, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2017, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39 (Suppl. S2), W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tang, J.; Di, R.; Liu, Q.; Wang, X.; Gan, S.; Zhang, X.; Zhang, J.; Hu, W.; Chu, M. Comparative Transcriptomics Reveal Key Sheep (Ovis aries) Hypothalamus LncRNAs that Affect Reproduction. Animals 2019, 9, 152. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, T.; Mano, A.; Shibasaki, T. Increased expression of miR-325-3p by urocortin 2 and its involvement in stress-induced suppression of LH secretion in rat pituitary. Am. J. Physiol. Metab. 2012, 302, E781–E787. [Google Scholar] [CrossRef]
- Lee, H.-J.; Palkovits, M.; Young, W.S. miR-7b, a microRNA up-regulated in the hypothalamus after chronic hyperosmolar stimulation, inhibits Fos translation. Proc. Natl. Acad. Sci. USA 2006, 103, 15669–15674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Han, M.; Zhou, Z.; Liu, Y.; He, X.; Jiang, Y.; Ouyang, Y.; Hong, Q.; Chu, M. Hypothalamic Transcriptome Analysis Reveals the Crucial MicroRNAs and mRNAs Affecting Litter Size in Goats. Front. Vet. Sci. 2021, 8, 747100. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cai, J.; Zhao, L.; Zhang, D.; Xu, G.; Hu, J.; Zhang, T.; Jin, M. NUMB suppression by miR-9-5P enhances CD44+ prostate cancer stem cell growth and metastasis. Sci. Rep. 2021, 11, 11210. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Fang, L.; Ouyang, B.; Zhang, X.; Zhan, S.; Feng, X.; Bai, Y.; Han, X.; Kim, H.; He, Q.; et al. Actl6a protects embryonic stem cells from differentiating into prim-itive endoderm. Stem Cells 2015, 33, 1782–1793. [Google Scholar] [CrossRef]
- Song, Y.; Mu, L.; Han, X.; Li, Q.; Dong, B.; Li, H.; Liu, X. MicroRNA-9 inhibits vasculogenic mimicry of glioma cell lines by suppress-ing Stathmin expression. J. Neurooncol. 2013, 115, 381–390. [Google Scholar] [CrossRef]
- Messina, A.; Langlet, F.; Chachlaki, K.; Roa, J.; Rasika, S.; Jouy, N.; Gallet, S.; Gaytan, F.; Parkash, J.; Tena-Sempere, M.; et al. A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat. Neurosci. 2016, 19, 835–844. [Google Scholar] [CrossRef]
- Roush, S.; Slack, F.J. The let-7 family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef]
- Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Hendrick, A.; Andrews, B.; Webster, N.; Murat, P.; Mach, P.; Brandi, R.; Robson, S.C.; et al. METTL1 Promotes let-7 microRNA pro-cessing via m7G methylation. Mol. Cell 2019, 74, 1278–1290.e9. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; He, K.; Ren, T.; Lou, Y.; Zhao, A. High-throughput sequencing reveals differential expression of miRNAs in prehierarchal follicles of laying and brooding geese. Physiol. Genom. 2016, 48, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, J.; Yang, Y.; Cheung, H.-H.A.; Chen, Z.; Chan, W.-Y. Conserved miR-10 family represses proliferation and induces apoptosis in ovarian granulosa cells. Sci. Rep. 2017, 7, 41304. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ye, Z.; Yang, H.; Xu, Q. Tailoring combinatorial lipid nanoparticles for intracellular delivery of nucleic acids, proteins, and drugs. Acta Pharm Sin B. 2022, 12, 2624–2639. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Du, X.; Liu, L.; Liu, H.; Pan, Z.; Li, Q. Upregulation of miR-146b promotes porcine ovarian granulosa cell apoptosis by at-tenuating CYP19A1. Domest. Anim. Endocrinol. 2021, 74, 106509. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.F.; Roser, J.F. Immunocytochemical localization of cytochrome P450 aromatase in the testis of prepubertal, pubertal, and postpubertal horses. Theriogenology 2003, 61, 293–299. [Google Scholar] [CrossRef]
- Sun, T.T.; Yang, Z.L.; Deng, J.X.; Zeng, L.H.; Jiang, H.S.; Yang, X.R. Cloning, sequence analysis and tissue expression profiling of CYP19A1 gene in Guangxi ma chicken. China Poultry 2017, 39, 60–62. [Google Scholar]
- Akçurin, S.; Türkkahraman, D.; Kim, W.-Y.; Durmaz, E.; Shin, J.-G.; Lee, S.-J. A Novel Null Mutation in P450 Aromatase Gene (CYP19A1) Associated with Development of Hypoplastic Ovaries in Humans. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 205–210. [Google Scholar] [CrossRef]
- Sytnyk, V.; Leshchyns’Ka, I.; Schachner, M. Neural Cell Adhesion Molecules of the Immunoglobulin Superfamily Regulate Synapse Formation, Maintenance, and Function. Trends Neurosci. 2017, 40, 295–308. [Google Scholar] [CrossRef]
- Parkash, J.; Kaur, G. Neuronal-glial plasticity in gonadotropin-releasing hormone release in adult female rats: Role of the pol-ysialylated form of the neural cell adhesion molecule. J. Endocrinol. 2005, 186, 397–409. [Google Scholar] [CrossRef]
- Rubinek, T.; Yu, R.; Hadani, M.; Barkai, G.; Nass, D.; Melmed, S.; Shimon, I. The cell adhesion molecules N-cadherin and neural cell ad-hesion molecule regulate human growth hormone: A novel mechanism for regulating pituitary hormone secretion. J. Clin. Endocrinol. Metab. 2003, 88, 3724–3730. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Gao, Y.; Lan, X.; Thomas, A.; Li, S. NCAM Mimetic Peptides: Potential Therapeutic Target for Neurological Disorders. Neurochem. Res. 2018, 43, 1714–1722. [Google Scholar] [CrossRef]
- Li, L.; Chen, H.; Wang, M.; Chen, F.; Gao, J.; Sun, S.; Li, Y.; Gao, D. NCAM-140 Translocation into Lipid Rafts Mediates the Neuroprotective Effects of GDNF. Mol. Neurobiol. 2016, 54, 2739–2751. [Google Scholar] [CrossRef]
- Li, H.T.; Qiu, T.; Chen, C.; Wang, C.Q. Research progress of connexin 43 in the field of osteoporosis. Chin. J. Difficult Complicat. Cases 2021, 20, 850–854. [Google Scholar]
- Miraoui, H.; Dwyer, A.; Pitteloud, N. Role of fibroblast growth factor (FGF) signaling in the neuroendocrine control of human reproduction. Mol. Cell. Endocrinol. 2011, 346, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.-C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Dai, Y.; Cui, F.; Pan, Y.; Zhang, H.; Xiao, J.; Fu, X. Expression of cytokines, growth factors and apoptosis-related signal mol-ecules in chronic pressure ulcer wounds healing. Spinal Cord. 2014, 52, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Saxena, P.; Pradhan, D.; Verma, R.; Kumar, S.N.; Deval, R.; Jain, A.K. Up-regulation of fibroblast growth factor receptor 1 due to prenatal tobacco exposure can lead to developmental defects in new born. J. Matern. Neonatal Med. 2018, 33, 1732–1743. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Q.; Xie, C.; Cai, Y.; Chen, X.; Hou, Y.; He, L.; Li, J.; Yao, M.; Chen, S.; et al. Peptide ligands targeting FGF receptors promote recovery from dorsal root crush injury via AKT/mTOR signaling. Theranostics 2021, 11, 10125–10147. [Google Scholar] [CrossRef]
- Tsai, P.S.; Moenter, S.M.; Postigo, H.R.; El Majdoubi, M.; Pak, T.R.; Gill, J.C.; Paruthiyil, S.; Werner, S.; Weiner, R.I. Targeted expression of a dominant-negative fibro-blast growth factor (FGF) receptor in gonadotropin-releasing hormone (GnRH) neurons reduces FGF responsiveness and the size of GnRH neuronal population. Mol. Endocrinol. 2005, 19, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Kunath, T.; Yamanaka, Y.; Detmar, J.; MacPhee, D.; Caniggia, I.; Rossant, J.; Jurisicova, A. Developmental differences in the expression of FGF receptors between human and mouse embryos. Placenta 2014, 35, 1079–1088. [Google Scholar] [CrossRef]
- Ozawa, M.; Yang, Q.E.; Ealy, A.D. The expression of fibroblast growth factor receptors during early bovine conceptus develop-ment and pharmacological analysis of their actions on trophoblast growth in vitro. Reproduction 2013, 145, 191–201. [Google Scholar] [CrossRef]
- Kang, M.; Garg, V.; Hadjantonakis, A.K. Lineage establishment and progression within the inner cell mass of the mouse blasto-cyst requires FGFR1 and FGFR2. Dev. Cell 2017, 41, 496–510.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.W.; Zhao, X.Y.; Zhang, Z.Y.; Hong, Y.; Fan, Y.; Ren, J.; Xing, Y. Fine Mapping of QTL and Identification of MEA1 as a Positional Candidate Gene Affecting Boar Reproduction Traits on SSC7. Acta Vet. Zootech. Sin. 2014, 45, 363–374. [Google Scholar]
- Tao, L.; He, X.Y.; Jiang, Y.T.; Lan, R.; Li, M.; Li, Z.M.; Yang, W.F.; Hong, Q.H.; Chu, M.X. Combined approaches to reveal genes associated with litter size in Yun-shang black goats. Anim. Genet. 2020, 51, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, K.G.; Segars, J.H. The role of adiponectin in reproduction: From polycystic ovary syndrome to assisted reproduction. Fertil. Steril. 2010, 94, 1949–1957. [Google Scholar] [CrossRef] [Green Version]
- Kubota, N.; Yano, W.; Kubota, T.; Yamauchi, T.; Itoh, S.; Kumagai, H.; Kozono, H.; Takamoto, I.; Okamoto, S.; Shiuchi, T.; et al. Adiponectin Stimulates AMP-Activated Protein Kinase in the Hypothalamus and Increases Food Intake. Cell Metab. 2007, 6, 55–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, I.; Sanchez-Alavez, M.; Tabarean, I.; Schaefer, J.; Holmberg, K.H.; Klaus, J.; Xia, F.; Marcondes, M.C.G.; Dubins, J.S.; Morrison, B.; et al. AdipoR1 and 2 are expressed on warm sen-sitive neurons of the hypothalamic preoptic area and contribute to central hyperthermic effects of adiponectin. Brain Res. 2011, 1423, 1–9. [Google Scholar] [CrossRef]
- Wu, Y.; Zhan, Z.; Quan, Y.; Yang, Y.; Chen, X.; Liu, L.; Wu, K.; Yu, M. SP1-mediated upregulation of LINGO-1 promotes degeneration of reti-nal ganglion cells in optic nerve injury. CNS Neurosci. Ther. 2020, 26, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, S.; Zeng, J.; Chen, M. miR-1271-5p inhibits cell proliferation and induces apoptosis in acute myeloid leukemia by targeting ZIC2. Mol. Med. Rep. 2018, 19, 508–514. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhou, Z.; He, X.; Tao, L.; Jiang, Y.; Lan, R.; Hong, Q.; Chu, M. Integrated analyses of miRNA-mRNA expression profiles of ovaries reveal the crucial interaction networks that regulate the prolificacy of goats in the follicular phase. BMC Genom. 2021, 22, 812. [Google Scholar] [CrossRef]
- Zimowska, G.; Shi, J.; Munguba, G.; Jackson, M.R.; Alpatov, R.; Simmons, M.N.; Shi, Y.; Sugrue, S.P. Pinin/DRS/memA Interacts with SRp75, SRm300 and SRrp130 in Corneal Epithelial Cells. Investig. Opthalmol. Vis. Sci. 2003, 44, 4715–4723. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guan, Q.; Nan, Y.; Ma, Q.; Zhong, Y. Overexpression of human MX2 gene suppresses cell proliferation, migration, and invasion via ERK/P38/NF-κB pathway in glioblastoma cells. J. Cell. Biochem. 2019, 120, 18762–18770. [Google Scholar] [CrossRef]
- Buragohain, L.; Kumar, R.; Nanda, T.; Phulia, S.; Mohanty, A.; Kumar, S.; Balhara, S.; Ghuman, S.; Singh, I.; Balhara, A. Serum MX2 Protein as Candidate Biomarker for Early Pregnancy Diagnosis in Buffalo. Reprod. Domest. Anim. 2016, 51, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, H.; Di, C.; Xie, Y.; Zhou, X.; Yan, J.; Zhao, Q. Comparative proteomic profiling and possible toxicological mechanism of acute injury induced by carbon ion radiation in pubertal mice testes. Reprod. Toxicol. 2015, 58, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Fortes, M.R.; Reverter, A.; Nagaraj, S.H.; Zhang, Y.; Jonsson, N.N.; Barris, W.; Lehnert, S.; Boe-Hansen, G.B.; Hawken, R.J. A single nucleotide polymorphism-derived regu-latory gene network underlying puberty in 2 tropical breeds of beef cattle. J. Anim. Sci. 2011, 89, 1669–1683. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequences (5′-3′) | Accession No. | Tm (°C) |
|---|---|---|---|
| NCAM1 | F: TGTGTGATGTGGTCAGCTCC | XM_018059775.1 | 60 |
| R: TGTGTGATGTGGTCAGCTCC | |||
| FGFR2 | F: ATGTCATCGTCGAGTACGCC | XM_018041428.1 | 60 |
| R: TAGGTGCACGACACCAAGTC | |||
| LINGO1 | F: CGCCTCAAGGTGTTGGAGAT | XM_018066520.1 | 60 |
| R: GACAGGGACGTCAGGTTGAG | |||
| MEA1 | F: AGGACGATGGCTGGCATAAG | XM_005696321.3 | 60 |
| R: CTTGGAGGGCCTTCTGTACC | |||
| SDCCAG8 | F: GGAGATTCTGGGGCAGTGTC | XM_013970102.2 | 60 |
| R: GTCACCTTCTCTCAGGGCAC | |||
| MX2 | F: CTCATTGACCTTCCCGGCAT | XM_018051644.1 | 60 |
| R: ATGGTCTCCTGCCTCTGGAT | |||
| RPL19 | F: ATCGCCAATGCCAACTC | XM_005693740.3 | 60 |
| R: CCTTTCGCTTACCTATACC | |||
| chi-miR-200a | CAGTAACACTGTCTGGTAACG | - | 60 |
| chi-let-7d-3p | GCAGCTATACGACCTGCT | - | 60 |
| chi-miR-10b-3p | CGCAGACAGATTCGATTC | - | 60 |
| chi-let-7b-3p | AGCTATACAACCTACTGCCTT | - | 60 |
| novel-miR-403 | GGGCCGGGCCT | - | 60 |
| novel-miR-1125 | GCCCCTGGGCCT | - | 60 |
| U6 snRNA | CAAGGATGACACGCAAATTCG | - | 60 |
| Items | Clean Reads | Mapped Reads | Mapping Ratio | Q20(%) | Q30(%) | GC Content (%) |
|---|---|---|---|---|---|---|
| LP-HF1 | 98,815,860 | 95,807,657 | 96.96% | 98.7;98.0 | 95.8;94.0 | 47.1;47.7 |
| LP-HF2 | 102,074,738 | 99,172,836 | 97.16% | 98.8;98.2 | 96.3;94.5 | 48.0;48.6 |
| LP-HF3 | 106,891,624 | 102,778,831 | 96.15% | 98.4;97.5 | 95.0;92.8 | 41.4;41.3 |
| LP-HF4 | 107,291,396 | 103,863,460 | 96.81% | 98.7;98.3 | 95.9;94.7 | 45.3;45.6 |
| LP-HF5 | 118,968,066 | 115,301,498 | 96.92% | 98.6;98.0 | 95.6;94.1 | 48.4;49.0 |
| LP-LF1 | 124,269,432 | 120,367,153 | 96.86% | 98.6;98.2 | 95.6;94.5 | 47.8;48.3 |
| LP-LF2 | 104,606,848 | 101,179,431 | 96.72% | 98.4;97.7 | 95.3;93.3 | 46.6;47.1 |
| LP-LF3 | 106,329,382 | 102,901,653 | 96.78% | 98.5;98.3 | 95.4;94.7 | 46.3;46.6 |
| LP-LF4 | 124,634,642 | 120,894,187 | 97.00% | 98.6;98.3 | 95.7;94.7 | 47.8;48.4 |
| LP-LF5 | 117,361,866 | 113,601,480 | 96.80% | 98.7;98.4 | 95.8;94.9 | 44.6;45.1 |
| Items | Raw Reads | Clean Reads | Clean Ratio | Mapped Reads | Mapping Ratio | Q20 | Q30 | GC Content |
|---|---|---|---|---|---|---|---|---|
| LP-HF1 | 24,366,316 | 20,116,645 | 82.56% | 19,590,837 | 97.39% | 99.29% | 97.69% | 50.31% |
| LP-HF2 | 21,343,198 | 13,008,981 | 60.95% | 12,775,878 | 98.21% | 99.18% | 97.55% | 50.95% |
| LP-HF3 | 26,925,011 | 26,264,285 | 97.55% | 25,677,239 | 97.76% | 99.40% | 97.89% | 47.71% |
| LP-HF4 | 20,392,398 | 18,804,252 | 92.21% | 18,460,000 | 98.17% | 99.40% | 97.87% | 48.68% |
| LP-HF5 | 23,659,871 | 14,275,718 | 60.34% | 13,988,821 | 97.99% | 98.98% | 97.19% | 50.67% |
| LP-LF1 | 28,524,952 | 25,571,497 | 89.65% | 24,371,807 | 95.31% | 99.25% | 97.27% | 51.79% |
| LP-LF2 | 27,742,510 | 26,491,767 | 95.49% | 25,784,762 | 97.33% | 99.09% | 96.69% | 48.59% |
| LP-LF3 | 27,171,386 | 24,975,767 | 91.92% | 24,300,180 | 97.30% | 99.10% | 96.60% | 48.52% |
| LP-LF4 | 27,578,592 | 24,944,377 | 90.45% | 24,450,755 | 98.02% | 99.32% | 97.67% | 48.82% |
| LP-LF5 | 21,960,009 | 21,033,490 | 95.78% | 20,562,709 | 97.76% | 99.16% | 97.40% | 48.27% |
| GO ID | GO_Term | Gene Name | q-Value |
|---|---|---|---|
| GO:0043168 | Anion binding | MX2, SEPT8, SNX17, RAB19, FES, RTEL1, ABCA3 | 0.0039 |
| GO:0036094 | Small molecule binding | MX2, SEPT8, IMPDH2, RAB19, FES, RTEL1, ABCA3 | 0.0041 |
| GO:0098590 | Plasma membrane region | ERC1, ADGRL1 | 0.0111 |
| GO:0060627 | Regulation of the vesicle-mediated transport | C1, TBC1D16 | 0.0118 |
| GO:0000166 | Nucleotide binding | MX2, SEPT8, IMPDH2, RAB19, FES, RTEL1, ABCA3 | 0.0118 |
| GO:1901265 | Nucleoside phosphate binding | MX2, SEPT8, IMPDH2, RAB19, FES, RTEL1, ABCA3 | 0.0118 |
| GO:0016043 | Cellular component organization | MX2, SUPT5H, SEMA4F, FLII, IMPDH2, ADGRL1, MRRF, MIEF2, C2, TBC1D16, FES, RTEL1, BCL9L, SDCCAG8 | 0.0118 |
| GO:0005543 | Phospholipid binding | SNX17, FES | 0.0118 |
| GO:0048284 | Organelle fusion | TBC1D16 | 0.0118 |
| GO:0071840 | Cellular component organization or biogenesis | MX2, SUPT5H, SEMA4F, FLII, IMPDH2, ADGRL1, MRRF, MIEF2, C2, TBC1D16, FES, RTEL1, BCL9L, SDCCAG8 | 0.0124 |
| GO:0017076 | Purine nucleotide binding | MX2, SEPT8, RAB19, FES, RTEL1, ABCA3 | 0.0141 |
| GO:0035639 | Purine ribonucleoside triphosphate binding | MX2, SEPT8, RAB19, FES, RTEL1, ABCA3 | 0.0164 |
| GO:0032555 | Purine ribonucleotide binding | MX2, SEPT8, RAB19, FES, RTEL1, ABCA3 | 0.0164 |
| GO:0051962 | Positive regulation of the nervous system development | HEYL, ADGRL1 | 0.0164 |
| GO:0016192 | Vesicle-mediated transport | SNX17, ERC1, C2, TBC1D16 | 0.0178 |
| GO:0032553 | Ribonucleotide binding | MX2, SEPT8, RAB19, FES, RTEL1, ABCA3 | 0.0178 |
| GO:0051179 | Localization | MX2, FXYD5, SNX17, ERC1, MIEF2, C2, SLC29A3, PLIN2, TBC1D16, SRGAP3, FES, CLN3, ABCA3 | 0.0183 |
| GO:0005488 | Binding | MX2, SEPT8, SUPT5H, EGFL8, SNX17, FLII, HEYL, ERC1, IMPDH2, ADGRL1, MRRF, C2, TBC1D16, SRGAP3, RTEL1, TEAD2, ABCA3 | 0.0183 |
| GO:0042734 | Presynaptic membrane | ERC1, ADGRL1 | 0.0203 |
| GO:0097367 | Carbohydrate derivative binding | MX2, SEPT8, RAB19, FES, RTEL1, ABCA3 | 0.0208 |
| GO:1901363 | Heterocyclic compound binding | MX2, SEPT8, SUPT5H, HEYL, IMPDH2, RAB19, FES, RTEL1, TEAD2, ABCA3 | 0.0212 |
| GO:0006810 | Transport | MX2, FXYD5, SNX17, ERC1, MIEF2, C2, SLC29A3, PLIN2, TBC1D16, CLN3, ABCA3 | 0.0213 |
| GO:0016050 | Vesicle organization | TBC1D16 | 0.0213 |
| GO:0045666 | Positive regulation of the neuron differentiation | HEYL | 0.0231 |
| GO:0046907 | Intracellular transport | MX2, SNX17, ERC1, MIEF2, TBC1D16 | 0.0231 |
| GO:0097060 | Synaptic membrane | ERC1, ADGRL1 | 0.0246 |
| GO:0045202 | Synapse | ERC1, ADGRL1 | 0.0261 |
| GO:0051234 | Establishment of the localization | MX2, FXYD5, SNX17, ERC1, MIEF2, C2, SLC29A3, PLIN2, TBC1D16, CLN3, ABCA3 | 0.0267 |
| GO:0044087 | Regulation of the cellular component biogenesis | MIEF2, FES | 0.0296 |
| GO:0051649 | Establishment of the localization in the cell | MX2, SNX17, ERC1, MIEF2, TBC1D16 | 0.0304 |
| GO:0097159 | Organic cyclic compound binding | MX2, SEPT8, SUPT5H, HEYL, IMPDH2, RAB19, FES, RTEL1, TEAD2, ABCA3 | 0.0311 |
| GO:0051960 | Regulation of the nervous system development | HEYL, ADGRL1 | 0.032368404 |
| GO:0035091 | Phosphatidylinositol binding | SNX17, FES | 0.0351 |
| GO:0044801 | Single-organism membrane fusion | TBC1D16 | 0.0371 |
| GO:0097458 | Neuron part | ERC1, ADGRL1, S100A1, MAPT | 0.0373 |
| GO:0043167 | Ion binding | MX2, SEPT8, EGFL8, SNX17, IMPDH2, C2, RAB19, FES, RTEL1, MAN2A2, ABCA3 | 0.0429 |
| GO:0051641 | Cellular localization | MX2, SNX17, ERC1, MIEF2, TBC1D16, CLN3 | 0.0457 |
| Gene Name | KEGG Pathway | Pathway ID |
|---|---|---|
| EVC | Hedgehog signaling pathway | ko04340 |
| EDEM3 | Protein processing in endoplasmic reticulum | ko04141 |
| SEMA4F | Axon guidance | ko04360 |
| DHDDS | Terpenoid backbone biosynthesis | ko00900 |
| EPS15L1 | Endocytosis | ko04144 |
| GP1BB | Platelet activation; Hematopoietic cell lineage; ECM-receptor interaction | ko04611; ko04640; ko04512 |
| ERC1 | NF-kappa B signaling pathway | ko04064 |
| IMPDH2 | Purine metabolism; Drug metabolism—other enzymes | ko00230; ko00983 |
| ERC1 | NF-kappa B signaling pathway | ko04064 |
| FLNC | MAPK signaling pathway; Focal adhesion | ko04010; ko04510 |
| ABCC5 | ABC transporters | ko02010 |
| C2 | Complement and coagulation cascades | ko04610 |
| PLIN2 | PPAR signaling pathway | ko03320 |
| ACSS2 | Carbon metabolism; Carbon fixation pathways in prokaryotes; Methane metabolism; Propanoate metabolism; Pyruvate metabolism; Glycolysis/Gluconeogenesis | ko01200; ko00720; ko00680; ko00640; ko00620; ko00010 |
| VAV2 | B cell receptor signaling pathway; Fc gamma R-mediated phagocytosis; Regulation of the actin cytoskeleton; Focal adhesion; cAMP signaling pathway; Chemokine signaling pathway; Fc epsilon RI signaling pathway; Leukocyte transendothelial migration; Natural killer cell mediated cytotoxicity; T cell receptor signaling pathway | ko04662; ko04666; ko04810; ko04510; ko04024; ko04062; ko04664; ko04670; ko04650; ko04660 |
| PLCB2 | Glutamatergic synapse; Phosphatidylinositol signaling system; Inositol phosphate metabolism; Renin secretion; cGMP—PKG signaling pathway; Wnt signaling pathway; Dopaminergic synapse; Long-term potentiation; Gap junction; Apelin signaling pathway; Calcium signaling pathway; Phospholipase D signaling pathway; Rap1 signaling pathway; Sphingolipid signaling pathway; Adrenergic signaling in cardiomyocytes; Vascular smooth muscle contraction; Gastric acid secretion; Pancreatic secretion; Salivary secretion; Aldosterone synthesis and secretion; Estrogen signaling pathway; Glucagon signaling pathway; GnRH signaling pathway; Insulin secretion; Melanogenesis; Oxytocin signaling pathway; Thyroid hormone signaling pathway; Thyroid hormone synthesis; Circadian entrainment; Endocrine and other factor-regulated calcium reabsorption; Chemokine signaling pathway; NOD-like receptor signaling pathway; Platelet activation; Cholinergic synapse; Long-term depression; Retrograde endocannabinoid signaling; Serotonergic synapse; Inflammatory mediator regulation of TRP channels; Phototransduction-fly | ko04724; ko04070; ko00562; ko04924; ko04022; ko04310; ko04728; ko04720; ko04540; ko04371; ko04020; ko04072; ko04015; ko04071; ko04261; ko04270; ko04971; ko04972; ko04970; ko04925; ko04915; ko04922; ko04912; ko04911; ko04916; ko04921; ko04919; ko04918; ko04713; ko04961; ko04062; ko04621; ko04611; ko04725; ko04730; ko04723; ko04726; ko04750; ko04745 |
| SRGAP3 | Axon guidance | ko04360 |
| FES | Axon guidance | ko04360 |
| EPS15L1 | Endocytosis | ko04144 |
| COL18A1 | Protein digestion and absorption | ko04974 |
| MAPK15 | IL-17 signaling pathway | ko04657 |
| COL11A2 | Protein digestion and absorption | ko04974 |
| CLN3 | Lysosome | ko04142 |
| COL13A1 | Protein digestion and absorption | ko04974 |
| TXNRD3 | Selenocompound metabolism | ko00450 |
| MAPK8IP3 | MAPK signaling pathway | ko04010 |
| MAN2A2 | N-Glycan biosynthesis; Various types of N-glycan biosynthesis | ko00510; ko00513 |
| MAPT | MAPK signaling pathway | ko04010 |
| SYVN1 | Ubiquitin mediated proteolysis; Protein processing in the endoplasmic reticulum | ko04120; ko04141 |
| TEAD2 | Hippo signaling pathway-multiple species; Hippo signaling pathway-fly; Hippo signaling pathway; MAPK signaling pathway-yeast | ko04392; ko04391; ko04390; ko04011 |
| MAPT | MAPK signaling pathway | ko04010 |
| L1CAM | Cell adhesion molecules (CAMs); Axon guidance | ko04514; ko04360 |
| ABCA3 | ABC transporters | ko02010 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, M.; Liang, C.; Liu, Y.; He, X.; Chu, M. Integrated Transcriptome Analysis Reveals the Crucial mRNAs and miRNAs Related to Fecundity in the Hypothalamus of Yunshang Black Goats during the Luteal Phase. Animals 2022, 12, 3397. https://doi.org/10.3390/ani12233397
Han M, Liang C, Liu Y, He X, Chu M. Integrated Transcriptome Analysis Reveals the Crucial mRNAs and miRNAs Related to Fecundity in the Hypothalamus of Yunshang Black Goats during the Luteal Phase. Animals. 2022; 12(23):3397. https://doi.org/10.3390/ani12233397
Chicago/Turabian StyleHan, Miaoceng, Chen Liang, Yufang Liu, Xiaoyun He, and Mingxing Chu. 2022. "Integrated Transcriptome Analysis Reveals the Crucial mRNAs and miRNAs Related to Fecundity in the Hypothalamus of Yunshang Black Goats during the Luteal Phase" Animals 12, no. 23: 3397. https://doi.org/10.3390/ani12233397