
Comprehensive Analyses of the Bacterial Population in Non-Healing Claw Lesions of Dairy Cattle
 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Isolation and Identification of Treponema spp.
2.3. Phylogenetic Tree Analysis
2.4. Specific PCR Assays
2.5. Immunohistochemical Staining
2.6. 16S rRNA Gene Amplicon-Based Metagenomic Analysis
2.7. Data Deposition
3. Results
3.1. Isolation of T. phagedenis from NHCLs
3.2. PCR Detection
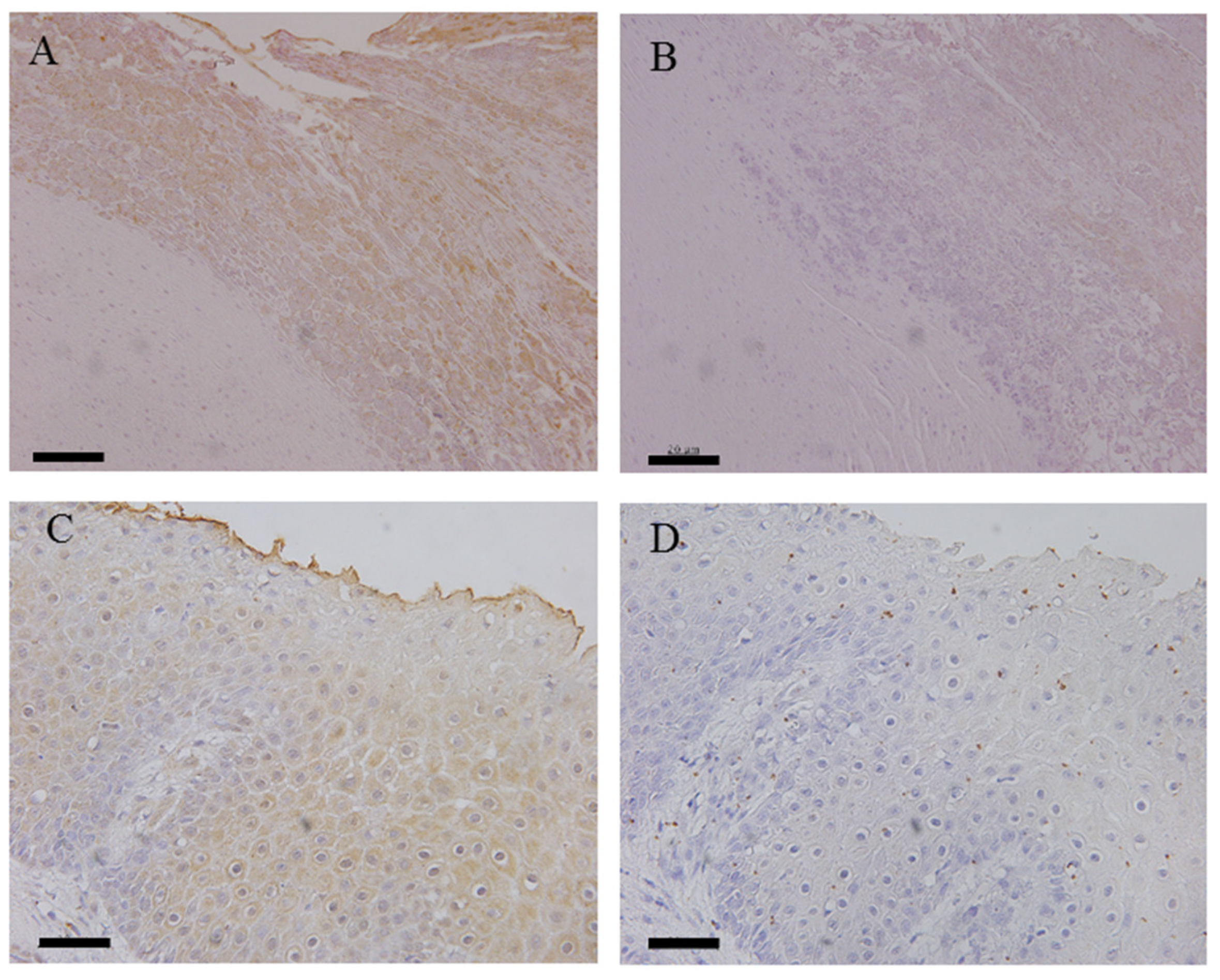
3.3. Immunohistochemical Analysis
3.4. Bacterial Populations in NHCL by 16S rRNA Gene Amplicon-Based Metagenomic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nir, O. What are production diseases, and how do we manage them? Acta Vet. Scand. Suppl. 2003, 44 (Suppl. 1), 21–32. [Google Scholar] [CrossRef]
- Evans, N.J.; Blowey, R.W.; Timofte, D.; Isherwood, D.R.; Brown, J.M.; Murray, R.; Paton, R.J.; Carter, S.D. Association between bovine digital dermatitis treponemes and a range of “non-healing” bovine hoof disorders. Vet. Rec. 2011, 168, 214. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.; Burgi, K. “Hairy Attack”: A new lesion affecting the corium of the abstract. In Proceedings of the 15th International Symposium & 7th Conference on Lameness in Ruminants, Kuopio, Finland, 13 June 2008; pp. 214–215. [Google Scholar]
- Holzhauer, M.; Vos, J.H. Claw health in Dutch dairy herds, an update of some recent disorders. Tijdschr. Diergeneeskd. 2009, 134, 200–205. [Google Scholar] [PubMed]
- Blowey, R.W. Non-healing hoof lesions in dairy cows. Vet. Rec. 2012, 32, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Blowey, R.W. Cattle Lameness and Hoofcare: An Illustrated Guide, 3rd ed.; 5m Publishing: Essex, UK, 2015; pp. 78–81. [Google Scholar]
- Staton, G.J.; Sullivan, L.E.; Blowey, R.W.; Carter, S.D.; Evans, N.J. Surveying bovine digital dermatitis and non-healing bovine foot lesions for the presence of Fusobacterium necrophorum, Porphyromonas endodontalis and Treponema pallidum. Vet. Rec. 2020, 186, 450. [Google Scholar] [CrossRef]
- Sykora, S.; Kofler, J.; Glonegger-Reichert, J.; Dietrich, J.; Auersperg, G.; Brandt, S. Treponema DNA in bovine “non-healing” versus common sole ulcers and white line disease. Vet. J. 2015, 205, 417–420. [Google Scholar] [CrossRef]
- Klitgaard, K.; Boye, M.; Capion, N.; Jensen, T.K. Evidence of multiple Treponema phylotypes involved in bovine digital dermatitis as shown by 16S rRNA gene analysis and fluorescence in situ hybridization. J. Clin. Microbiol. 2008, 46, 3012–3020. [Google Scholar] [CrossRef]
- Hori, K.; Ooshita, K.; Suzuki, N.; Kaneko, S.; Ichiba, S.; Ito, T.; Katayama, T.; Morita, T.; Sunden, Y.; Tsuka, T. A case study of serious pedal osteitis which is thought to be caused by spirillum in a Jersey cow for milk production. Hiroshima J. Vet. Med. 2016, 31, 11–14. Available online: http://www.hiro-vet.or.jp/_src/97410/p011-014.pdf (accessed on 8 November 2022). (In Japanese (Abstract in English)).
- Nordhoff, M.; Moter, A.; Schrank, K.; Wieler, L.H. High prevalence of treponemes in bovine digital dermatitis-A molecular epidemiology. Vet. Microbiol. 2008, 131, 293–300. [Google Scholar] [CrossRef]
- Read, D.H.; Walker, R.L.; Castro, A.E.; Sundbergh, J.P.; Thurmond, M.C. An invasive spirochaete associated with interdigital papillomatosis of dairy cattle. Vet. Rec. 1992, 130, 59–60. [Google Scholar] [CrossRef]
- Ohya, T.; Yamaguchi, H.; Nii, Y.; Ito, H. Isolation of Campylobacter sputorum from lesions of papillomatous digital dermatitis in dairy cattle. Vet. Rec. 1999, 145, 316–318. [Google Scholar] [CrossRef]
- Schroeder, C.M.; Parlor, K.W.; Marsh, T.L.; Ames, N.K.; Goeman, A.K.; Walker, R.D. Characterization of the predominant anaerobic bacterium recovered from digital dermatitis lesions in three Michigan dairy cows. Anaerobe 2003, 9, 151–155. [Google Scholar] [CrossRef]
- Warnick, L.D.; Nydam, D.; Maciel, A.; Guard, C.L.; Wade, S.E. Udder cleft dermatitis and sarcoptic mange in a dairy herd. J. Am. Vet. Med. Assoc. 2002, 221, 273–276. [Google Scholar] [CrossRef]
- Wyss, C.; Dewhirst, F.E.; Paster, B.J.; Thurnheer, T.; Luginbühl, A. Guggenheimella bovis gen. nov., sp. nov., isolated from lesions of bovine dermatitis digitalis. Int. J. Syst. Evol. Microbiol. 2005, 55, 667–671. [Google Scholar] [CrossRef]
- Yano, T.; Moe, K.K.; Yamazaki, K.; Ooka, T.; Hayashi, T.; Misawa, N. Identification of candidate pathogens of papillomatous digital dermatitis in dairy cattle from quantitative 16S rRNA clonal analysis. Vet. Microbiol. 2010, 143, 352–362. [Google Scholar] [CrossRef]
- Gotoh, Y.; Chiba, K.; Sekiyama, Y.; Okada, K.; Hayashi, T.; Misawa, N. 16S rRNA-based amplicon analysis of changes in the bacterial population in the lesions of papillomatous digital dermatitis in dairy cattle after topical treatment with allyl isothiocyanate. Microbiol. Immunol. 2020, 64, 416–423. [Google Scholar] [CrossRef]
- Mitsuoka, T.; Sega, T.; Yamamoto, S. Improved methodology of qualitative and quantitative analysis of the intestinal flora of man and animals. Zent. Bakteriol. Orig. 1965, 195, 455–469. [Google Scholar]
- Yano, T.; Yamagami, R.; Misumi, K.; Kubota, C.; Moe, K.K.; Hayashi, T.; Yoshitani, K.; Ohtake, O.; Misawa, N. Genetic heterogeneity among strains of Treponema phagedenis-like spirochetes isolated from dairy cattle with papillomatous digital dermatitis in Japan. J. Clin. Microbiol. 2009, 47, 727–733. [Google Scholar] [CrossRef]
- Misawa, N.; Kawashima, K.; Kawamoto, H.; Kondo, F. Development of a combined filtration-enrichment culture followed by a one-step duplex PCR technique for the rapid detection of Campylobacter jejuni and C. coli in human faecal samples. J. Med. Microbiol. 2002, 51, 86–89. [Google Scholar] [CrossRef]
- Edwards, U.; Rogall, T.; Blöcker, H.; Emde, M.; Böttger, E.C. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 1989, 17, 7843–7853. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinforma. 2002, 2, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Beninger, C.; Naqvi, S.A.; Naushad, S.; Orsel, K.; Luby, C.; Derakhshani, H.; Khafipour, E.; De Buck, J. Associations between digital dermatitis lesion grades in dairy cattle and the quantities of four Treponema species. Vet. Res. 2018, 49. [Google Scholar] [CrossRef] [PubMed]
- Asai, Y.; Jinno, T.; Igarashi, H.; Ohyama, Y.; Ogawa, T. Detection and quantification of oral treponemes in subgingival plaque by real-time PCR. J. Clin. Microbiol. 2002, 40, 3334–3340. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.; Hickford, J.; Sedcole, R.; Zhou, H. Dichelobacter nodosus, Fusobacterium necrophorum and the epidemiology of footrot. Anaerobe 2009, 15, 173–176. [Google Scholar] [CrossRef]
- Moe, K.K.; Yano, T.; Misumi, K.; Kubota, C.; Yamazaki, W.; Muguruma, M.; Misawa, N. Analysis of the IgG immune response to Treponema phagedenis-like spirochetes in individual dairy cattle with papillomatous digital dermatitis. Clin. Vaccine Immunol. 2010, 17, 376–383. [Google Scholar] [CrossRef]
- Evan, B.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “all-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, 643–648. [Google Scholar] [CrossRef]
- Brogden, K.A. Polymicrobial diseases of animals and humans. In Polymicrobial Diseases; Brogden, K.A., Guthmiller, J.M., Eds.; ASM Press: Washington, DC, USA, 2002; Chapter 1. [Google Scholar]
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Prim. 2017, 3, 17038. [Google Scholar] [CrossRef]
- Newsome, R.F.; Green, M.J.; Bell, N.J.; Bollard, N.J.; Mason, C.S.; Whay, H.R.; Huxley, J.N. A prospective cohort study of digital cushion and corium thickness. Part 2: Does thinning of the digital cushion and corium lead to lameness and claw horn disruption lesions? J. Dairy Sci. 2017, 100, 4759–4771. [Google Scholar] [CrossRef]
- Bay, V.; Griffiths, B.; Carter, S.; Evans, N.J.; Lenzi, L.; Bicalho, R.C.; Oikonomou, G. 16S rRNA amplicon sequencing reveals a polymicrobial nature of complicated claw horn disruption lesions and interdigital phlegmon in dairy cattle. Sci. Rep. 2018, 8, 15529. [Google Scholar] [CrossRef]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum-symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Socransky, S.S. Criteria for the infectious agents in dental caries and periodontal disease. J. Clin. Periodontol. 1979, 6, 16–21. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
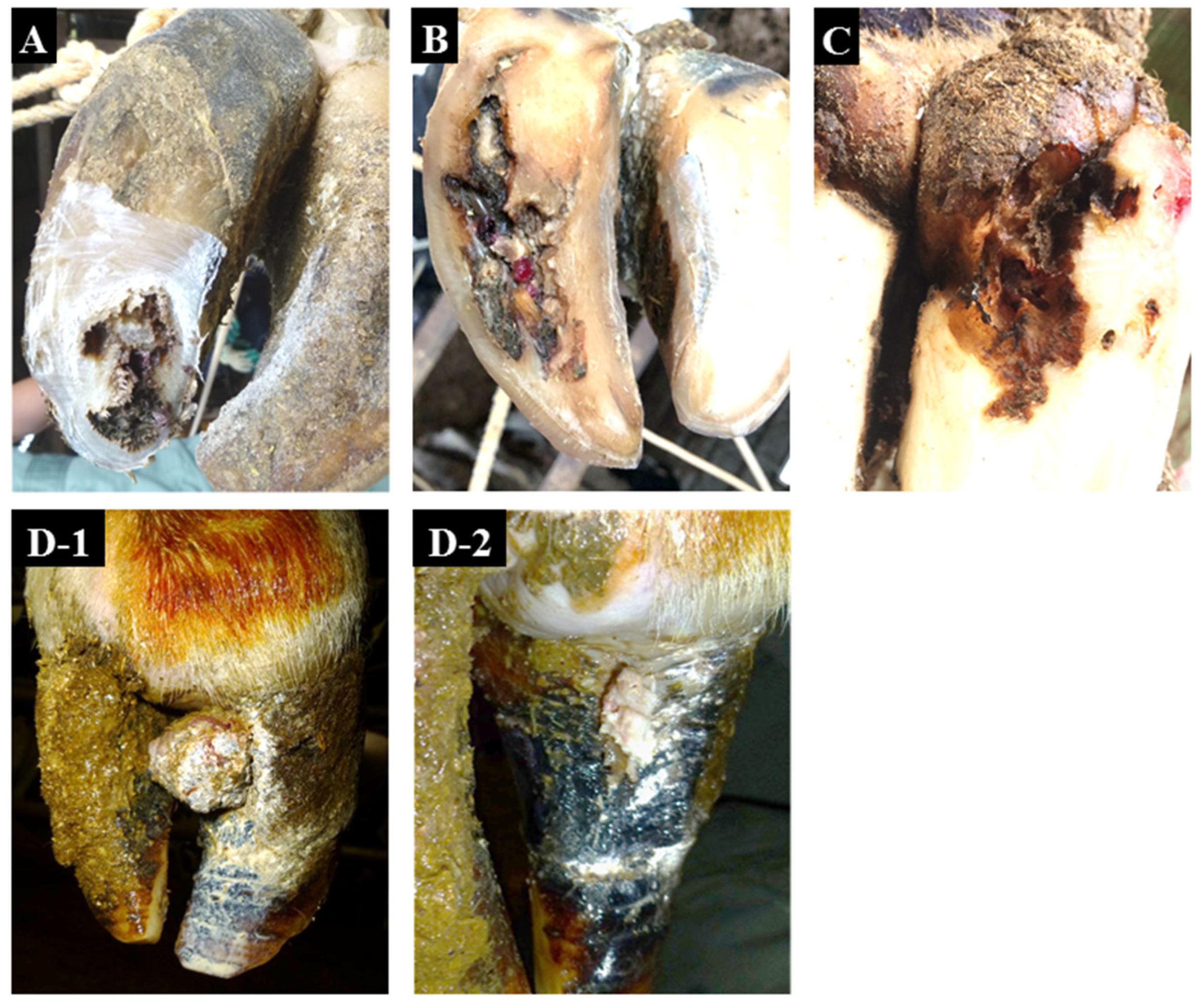
| Condition | Affected Limb/Claw | Zones Affected | Characteristic Signs | Common Signs |
|---|---|---|---|---|
| toe necrosis (TN) | all limb, lateral/medial claw | sole at the toe | necrosis progresses from the hoof horn at the toe to the deeper dermis and bone | distinctive odor |
| non-healing white line disease (nhWLD) | all limb, lateral/medial claw | white line | necrosis progresses from the white line to a deeper level which spreads to the surrounding area and extends to the bone | distinctive odor |
| non-healing sole ulcer (nhSU) | all limb, lateral/medial claw | sole–heel junction | necrosis progresses from the sole–heel junction to a deeper level which spreads to the surrounding area and extends to the bone | distinctive odor |
| non-healing verrucous-like lesions (nhVLL) | all limb, lateral/medial claw | dorsal hoof wall | verrucous-like lesions occur in the dermis within the dorsal hoof wall and appear by lacerating the dorsal hoof wall | distinctive odor |
| Sample No. | Farm | Lesion Type | Isolation | PCR | Antigen | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| T. phagedenis | T. phagedenis | T. denticola | T. medium | T. pedis | F. necrophorum | T. phagedenis | F. necrophorum | |||
| 1 | A | TN | + | + | - | + | + | + | + | - |
| 2 | B | TN | - | + | - | - | + | + | + | + |
| 3 | C | TN | - | + | + | + | + | + | + | + |
| 4 | C | nhWLD | - | + | + | + | + | + | + | + |
| 5 | F | nhWLD | - | + | + | + | + | + | + | + |
| 6 | G | nhWLD | - | + | - | - | + | + | - | + |
| 7 * | C | nhSU | + | - | - | - | + | + | + | + |
| 8 * | C | nhSU | - | - | - | - | - | - | - | + |
| 9 | C | nhSU | - | + | - | - | + | + | ND | + |
| 10 | G | nhSU | - | - | - | - | - | + | - | - |
| 11 | B | nhVLL | ND | + | + | + | + | + | + | + |
| 12 | B | nhVLL | - | + | + | + | + | + | + | + |
| 13 | D | Control | - | - | - | - | - | - | - | - |
| 14 | D | Control | - | - | - | - | - | - | - | - |
| 15 | E | Control | - | - | - | - | - | - | - | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hori, K.; Taniguchi, T.; Elpita, T.; Khemgaew, R.; Sasaki, S.; Gotoh, Y.; Yasutomi, I.; Misawa, N. Comprehensive Analyses of the Bacterial Population in Non-Healing Claw Lesions of Dairy Cattle. Animals 2022, 12, 3584. https://doi.org/10.3390/ani12243584
Hori K, Taniguchi T, Elpita T, Khemgaew R, Sasaki S, Gotoh Y, Yasutomi I, Misawa N. Comprehensive Analyses of the Bacterial Population in Non-Healing Claw Lesions of Dairy Cattle. Animals. 2022; 12(24):3584. https://doi.org/10.3390/ani12243584
Chicago/Turabian StyleHori, Kaoru, Takako Taniguchi, Trigan Elpita, Rathanon Khemgaew, Satomi Sasaki, Yasuhiro Gotoh, Ichiro Yasutomi, and Naoaki Misawa. 2022. "Comprehensive Analyses of the Bacterial Population in Non-Healing Claw Lesions of Dairy Cattle" Animals 12, no. 24: 3584. https://doi.org/10.3390/ani12243584
APA StyleHori, K., Taniguchi, T., Elpita, T., Khemgaew, R., Sasaki, S., Gotoh, Y., Yasutomi, I., & Misawa, N. (2022). Comprehensive Analyses of the Bacterial Population in Non-Healing Claw Lesions of Dairy Cattle. Animals, 12(24), 3584. https://doi.org/10.3390/ani12243584