
Mutation Characteristics and Phylogenetic Analysis of Five Leishmania Clinical Isolates

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Parasites
2.2. Leishmania Culture and Acquisition of Genomic DNA
2.3. Whole-Genome Resequencing and Analysis
2.3.1. Reference Genome
2.3.2. Variant Detection and Annotation
2.4. Genome Assembly and Phylogenetic Analysis
2.5. Mutation Analysis in Virulence and Resistance Associated Genes
3. Results
3.1. Resequencing Analysis
3.1.1. Mapping Results with Reference Genome
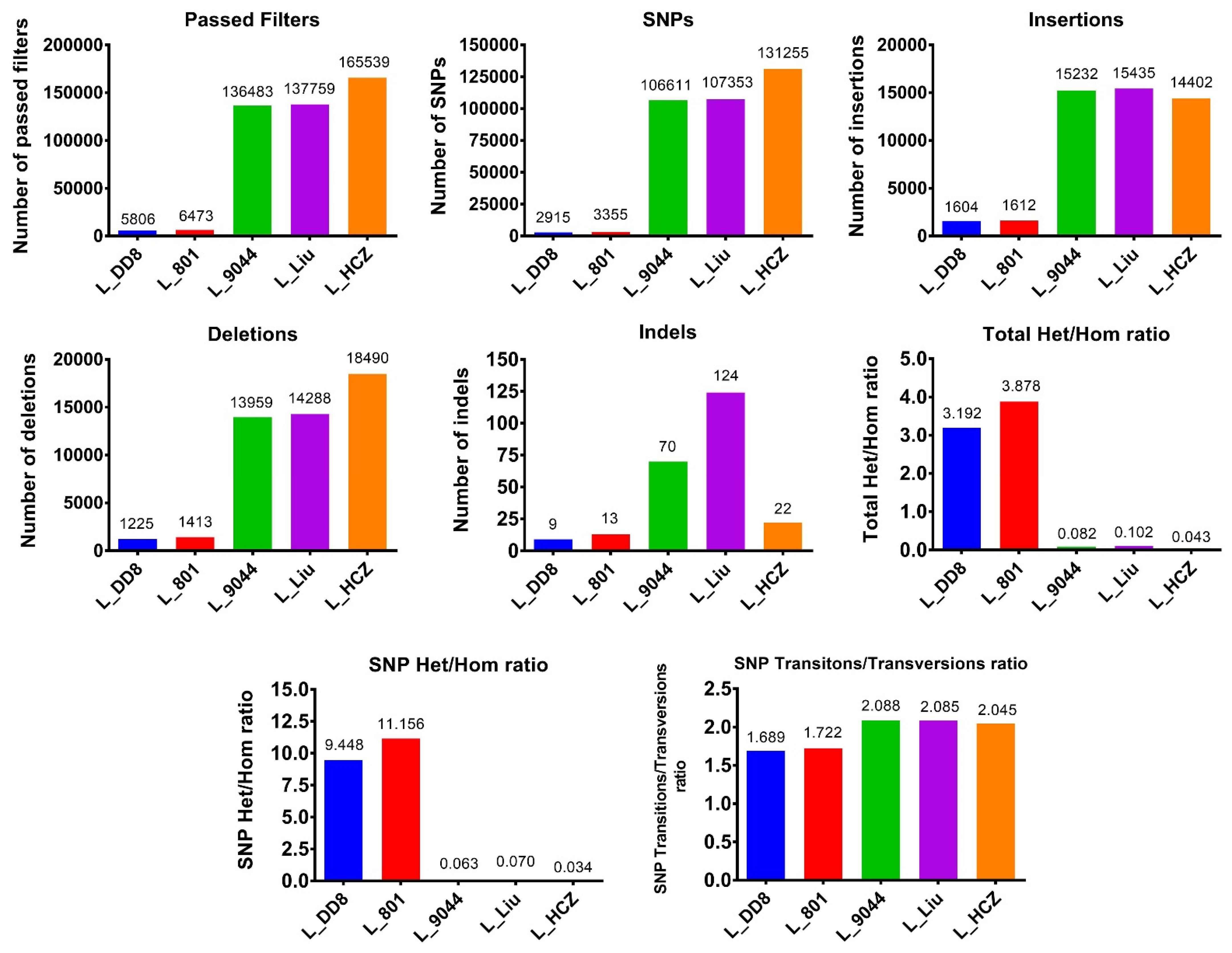
3.1.2. Detection Results of SNPs and InDels
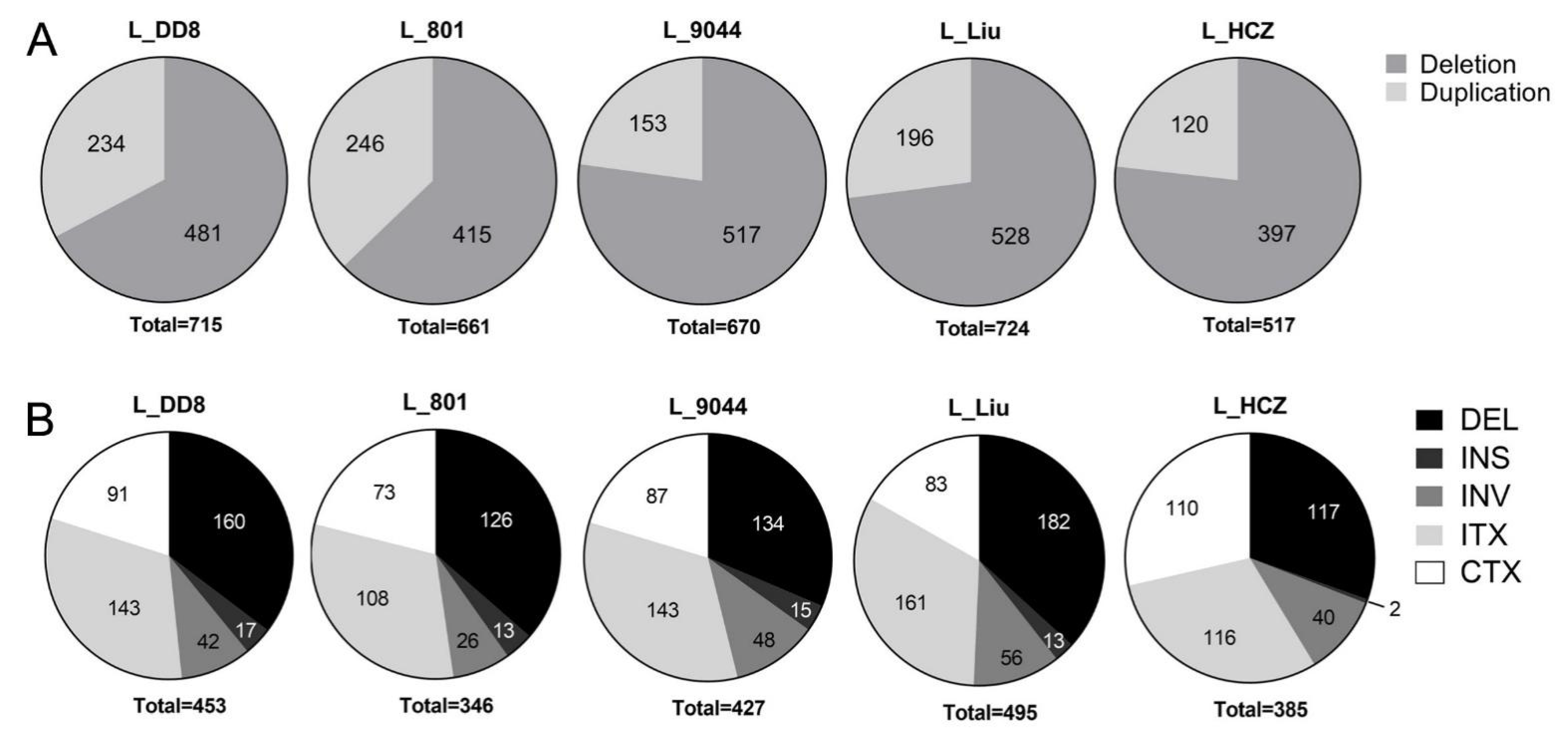
3.1.3. Detection Results of CNV and SV
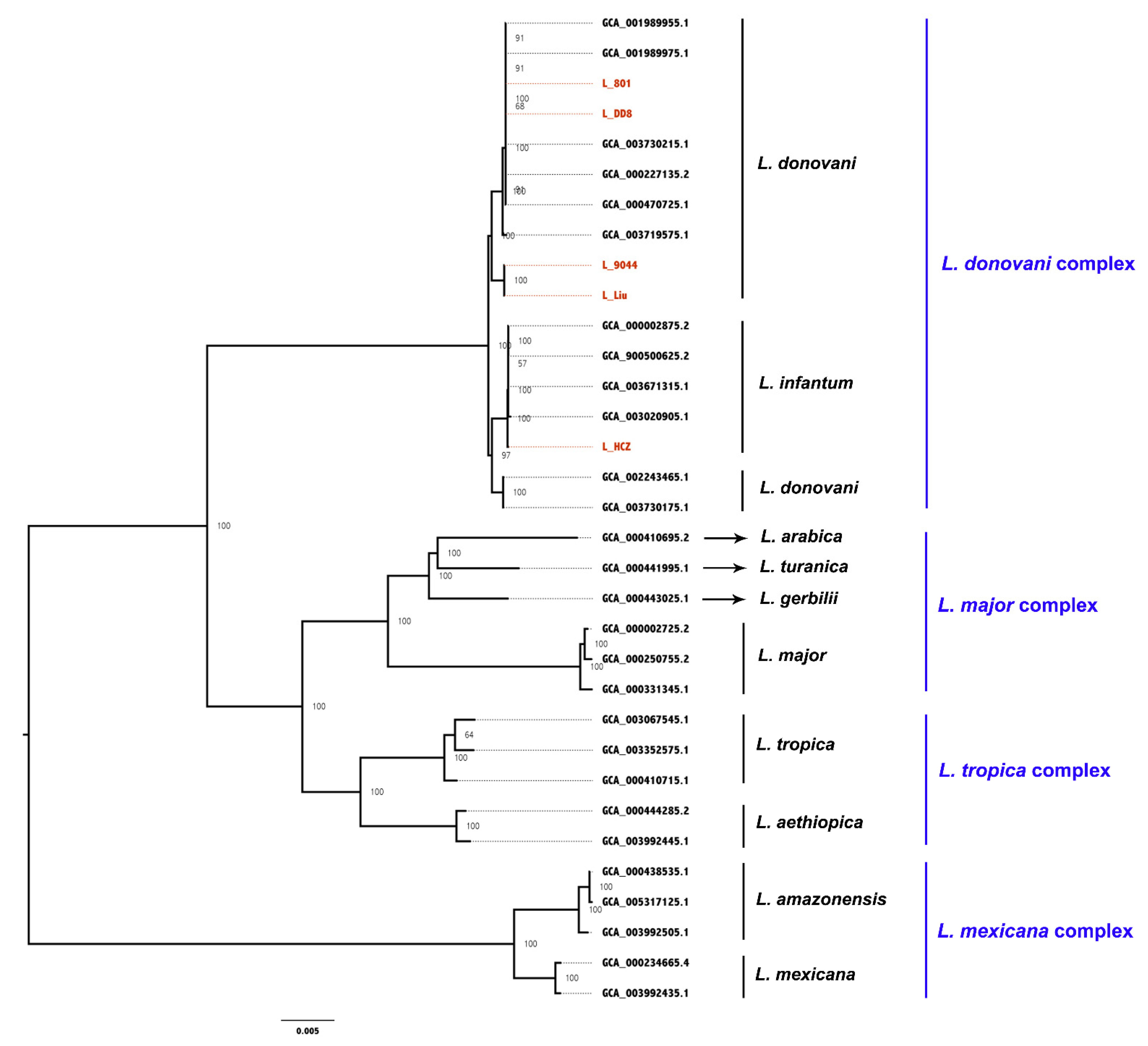
3.2. Whole-Gene Phylogenetic Analysis
3.3. Mutation Analysis of L_HCZ
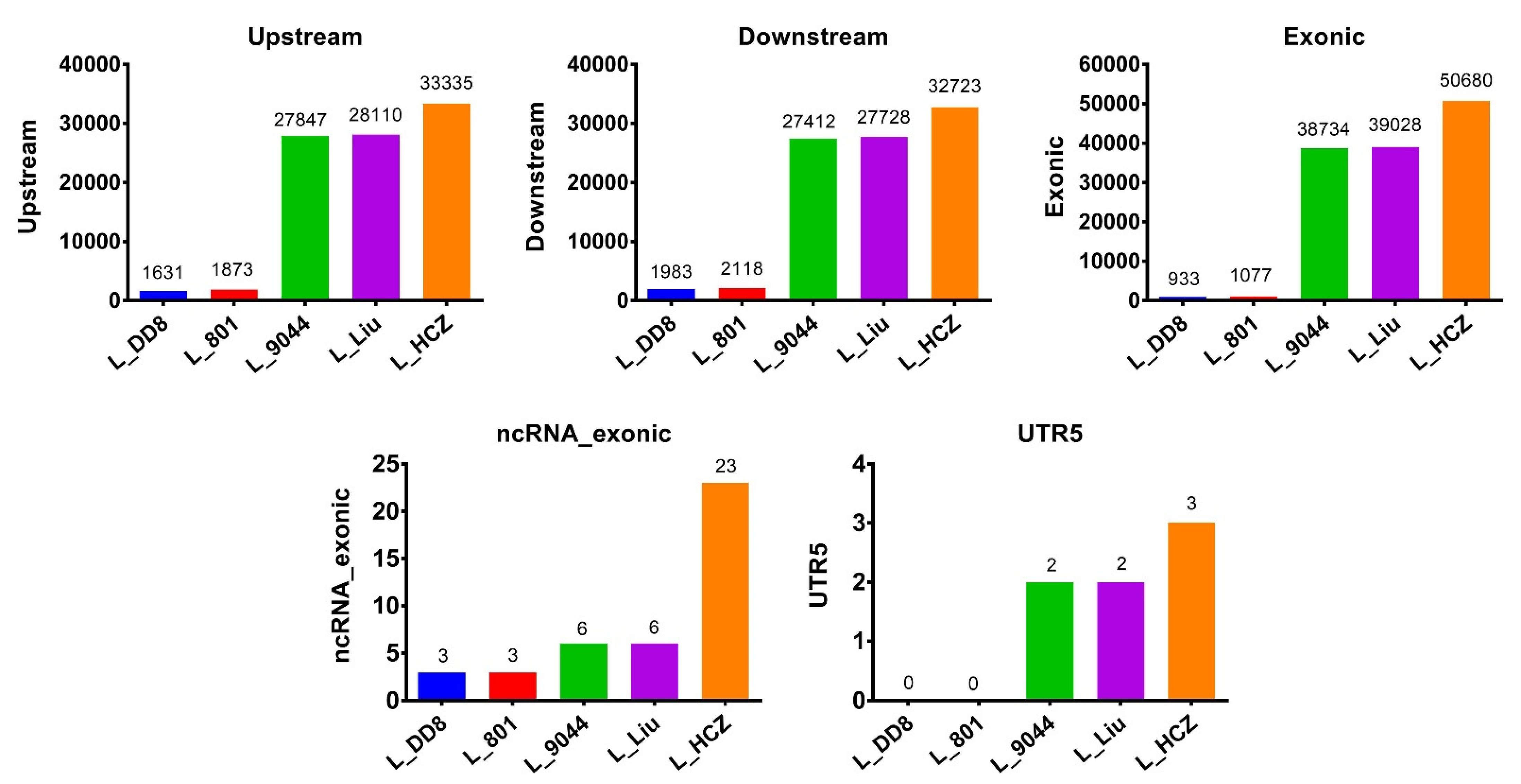
3.3.1. Exon Mutations in Five Leishmania Isolates
3.3.2. Mutation Analysis in Nine Genes Associated with Antimony Resistance
3.3.3. Mutation Analysis in Nine Genes Associated with Virulence
3.3.4. Mutation Analysis of 18 Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guerin, P.J.; Olliaro, P.; Sundar, S.; Boelaert, M.; Croft, S.L.; Desjeux, P.; Wasunna, M.K.; Bryceson, A.D. Visceral leishmaniasis: Current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect. Dis. 2002, 2, 494–501. [Google Scholar] [CrossRef]
- Hide, M.; Bras-Gonçalves, R.; Bañuls, A.L. Specific cpb copies within the Leishmania donovani complex: Evolutionary interpretations and potential clinical implications in humans. Parasitology 2007, 134, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostygov, A.Y.; Karnkowska, A.; Votýpka, J.; Tashyreva, D.; Maciszewski, K.; Yurchenko, V.; Lukeš, J. Euglenozoa: Taxonomy, diversity and ecology, symbioses and viruses. Open Biol. 2021, 11, 200407. [Google Scholar] [CrossRef] [PubMed]
- Maslov, D.A.; Opperdoes, F.R.; Kostygov, A.Y.; Hashimi, H.; Lukeš, J.; Yurchenko, V. Recent advances in trypanosomatid research: Genome organization, expression, metabolism, taxonomy and evolution. Parasitology 2019, 146, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votýpka, J.; Marty, P.; Delaunay, P.; Sereno, D. A historical overview of the classification, evolution, and dispersion of Leishmania parasites and sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef]
- Fraga, J.; Montalvo, A.M.; De Doncker, S.; Dujardin, J.C.; Van der Auwera, G. Phylogeny of Leishmania species based on the heat-shock protein 70 gene. Infect. Genet. Evol. 2010, 10, 238–245. [Google Scholar] [CrossRef]
- Akhoundi, M.; Downing, T.; Votýpka, J.; Kuhls, K.; Lukeš, J.; Cannet, A.; Ravel, C.; Marty, P.; Delaunay, P.; Kasbari, M.; et al. Leishmania infections: Molecular targets and diagnosis. Mol. Asp. Med. 2017, 57, 1–29. [Google Scholar] [CrossRef]
- Cantacessi, C.; Dantas-Torres, F.; Nolan, M.J.; Otranto, D. The past, present, and future of Leishmania genomics and transcriptomics. Trends Parasitol. 2015, 31, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Yurchenko, V.; Butenko, A.; Kostygov, A.Y. Genomics of trypanosomatidae: Where we stand and what needs to be done? Pathogens 2021, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- Real, F.; Vidal, R.O.; Carazzolle, M.F.; Mondego, J.M.; Costa, G.G.; Herai, R.H.; Würtele, M.; de Carvalho, L.M.; Carmona e Ferreira, R.; Mortara, R.A.; et al. The genome sequence of Leishmania (Leishmania) amazonensis: Functional annotation and extended analysis of gene models. DNA Res. 2013, 20, 567–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wincker, P.; Ravel, C.; Blaineau, C.; Pages, M.; Jaufrett, Y.; Dedet, J.P.; Bastien, P. The Leishmania genome comprises 36 chromosomes conserved across widely divergent human pathogenic species. Nucleic Acids Res. 1998, 24, 1688–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.A.; Adlem, E.; Aert, R.; et al. The genome of the kinetoplastid parasite, Leishmania major. Science 2005, 309, 436–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardley, V.; Croft, S.L.; De Doncker, S.; Dujardin, J.C.; Koirala, S.; Rijal, S.; Miranda, C.; Llanos-Cuentas, A.; Chappuis, F. The sensitivity of clinical isolates of Leishmania from Peru and Nepal to miltefosine. Am. J. Trop. Med. Hyg. 2005, 73, 272–275. [Google Scholar] [CrossRef]
- Sánchez-Cañete, M.P.; Carvalho, L.; Pérez-Victoria, F.J.; Gamarro, F.; Castanys, S. Low plasma membrane expression of the miltefosine transport complex renders Leishmania braziliensis refractory to the drug. Antimicrob. Agents Chemother. 2009, 53, 1305–1313. [Google Scholar] [CrossRef] [Green Version]
- Horn, D.; Duraisingh, M.T. Antiparasitic chemotherapy: From genomes to mechanisms. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 71–94. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Kosoy, M.; Dittmar, K. HGTector: An automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genom. 2014, 15, 717. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 198–203. [Google Scholar] [CrossRef]
- He, J.; Huang, F.; Zhang, J.; Chen, H.; Chen, Q.; Zhang, J.; Li, J.; Zheng, Z.; Chen, D.; Chen, J. DNA prime-protein boost vaccine encoding HLA-A2, HLA-A24 and HLA-DR1 restricted epitopes of CaNA2 against visceral leishmaniasis. Immunology 2019, 156, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009, 6, 677–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Fernández, A.; Gómez, S. Solving non-uniqueness in agglomerative hierarchical clustering using Multidendrograms. J. Classif. 2008, 25, 43–65. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dozmorov, M.G. GitHub statistics as a measure of the impact of open-source bioinformatics software. Front. Bioeng. Biotechnol. 2018, 6, 198. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Leprohon, P.; Fernandez-Prada, C.; Gazanion, É.; Monte-Neto, R.; Ouellette, M. Drug resistance analysis by next generation sequencing in Leishmania. Int. J. Parasitol. Drugs Drug Resist. 2014, 5, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Mohebali, M.; Kazemirad, E.; Hajjaran, H.; Kazemirad, E.; Oshaghi, M.A.; Raoofian, R.; Teimouri, A. Gene expression analysis of antimony resistance in Leishmania tropica using quantitative real-time PCR focused on genes involved in trypanothione metabolism and drug transport. Arch. Dermatol. Res. 2019, 311, 9–17. [Google Scholar] [CrossRef]
- Bifeld, E.; Clos, J. The genetics of Leishmania virulence. Med. Microbiol. Immunol. 2015, 204, 619–634. [Google Scholar] [CrossRef]
- Lukes, J.; Mauricio, I.L.; Schönian, G.; Dujardin, J.C.; Soteriadou, K.; Dedet, J.P.; Kuhls, K.; Tintaya, K.W.; Jirků, M.; Chocholová, E.; et al. Evolutionary and geographical history of the Leishmania donovani complex with a revision of current taxonomy. Proc. Natl. Acad. Sci. USA 2007, 104, 9375–9380. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A.; Lahav, T.; Holland, N.; Aggarwal, G.; Anupama, A.; Huang, Y.; Volpin, H.; Myler, P.J.; Zilberstein, D. Analysis of the Leishmania donovani transcriptome reveals an ordered progression of transient and permanent changes in gene expression during differentiation. Mol. Biochem. Parasitol. 2007, 152, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuse, S.; Fleurie, A.; Zucchini, L.; Lesterlin, C.; Grangeasse, C. Role of eukaryotic-like serine/threonine kinases in bacterial cell division and morphogenesis. FEMS Microbiol. Rev. 2016, 40, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quek, H.; Lim, Y.C.; Lavin, M.F.; Roberts, T.L. PIKKing a way to regulate inflammation. Immunol. Cell Biol. 2018, 96, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Saada, E.A.; Kabututu, Z.P.; Lopez, M.; Shimogawa, M.M.; Langousis, G.; Oberholzer, M.; Riestra, A.; Jonsson, Z.O.; Wohlschlegel, J.A.; Hill, K.L. Insect stage-specific receptor adenylate cyclases are localized to distinct subdomains of the Trypanosoma brucei Flagellar membrane. Eukaryot. Cell 2014, 13, 1064–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, A.; Sharma, P.; Redhu, N.S.; Singh, S. Kinesin motor domain of Leishmania donovani as a future vaccine candidate. Clin. Vaccine Immunol. 2008, 15, 836–842. [Google Scholar] [CrossRef] [Green Version]
- Silva-Almeida, M.; Pereira, B.A.; Ribeiro-Guimarães, M.L.; Alves, C.R. Proteinases as virulence factors in Leishmania spp. infection in mammals. Parasites Vectors 2012, 5, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.P.; McGwire, B.S. Molecular determinants and regulation of Leishmania virulence. Kinetoplastid Biol. Dis. 2002, 1, 1. [Google Scholar] [CrossRef]
- Rochette, A.; McNicoll, F.; Girard, J.; Breton, M.; Leblanc, E.; Bergeron, M.G.; Papadopoulou, B. Characterization and developmental gene regulation of a large gene family encoding amastin surface proteins in Leishmania spp. Mol. Biochem. Parasitol. 2005, 140, 205–220. [Google Scholar] [CrossRef]
- Asai, D.J.; Wilkes, D.E. The dynein heavy chain family. J. Eukaryot. Microbiol. 2004, 51, 23–29. [Google Scholar] [CrossRef]
- Clayton, C.E. Gene expression in Kinetoplastids. Curr. Opin. Microbiol. 2016, 32, 46–51. [Google Scholar] [CrossRef]
- Sommer, A.; Christensen, E.; Schwenger, S.; Seul, R.; Haas, D.; Olbrich, H.; Omran, H.; Sass, J.O. The molecular basis of aminoacylase 1 deficiency. Biochim. Biophys. Acta. 2011, 1812, 685–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvage, V.; Aubert, D.; Escotte-Binet, S.; Villena, I. The role of ATP-binding cassette (ABC) proteins in protozoan parasites. Mol. Biochem. Parasitol. 2009, 167, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Ouameur, A.A.; Girard, I.; Légaré, D.; Ouellette, M. Functional analysis and complex gene rearrangements of the folate/biopterin transporter (FBT) gene family in the protozoan parasite Leishmania. Mol. Biochem. Parasitol. 2008, 162, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Downing, T.; Imamura, H.; Decuypere, S.; Clark, T.G.; Coombs, G.H.; Cotton, J.A.; Hilley, J.D.; de Doncker, S.; Maes, L.; Mottram, J.C.; et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011, 21, 2143–2156. [Google Scholar] [CrossRef] [Green Version]
- Victoir, K.; Dujardin, J.C. How to succeed in parasitic life without sex? Asking Leishmania. Trends Parasitol. 2002, 18, 81–85. [Google Scholar] [CrossRef]
- McDonagh, P.D.; Myler, P.J.; Stuart, K. The unusual gene organization of Leishmania major chromosome 1 may reflect novel transcription processes. Nucleic Acids Res. 2000, 28, 2800–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downing, T.; Stark, O.; Vanaerschot, M.; Imamura, H.; Sanders, M.; Decuypere, S.; de Doncker, S.; Maes, I.; Rijal, S.; Sunder, S.; et al. Genome-wide SNP and microsatellite variation illuminate population-level epidemiology in the Leishmania donovani species complex. Infect. Genet. Evol. 2012, 12, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Y.; Lu, X.J.; Du, X.Q.; Jian, J.; Shu, L.; Ma, Y. Phylogenetic and evolutionary analysis of Chinese Leishmania isolates based on multilocus sequence typing. PLoS ONE 2013, 8, e63124. [Google Scholar] [CrossRef] [Green Version]
- Guan, L.R.; Yang, Y.Q.; Qu, J.Q.; Ren, H.Y.; Chai, J.J. Discovery and study of cutaneous leishmaniasis in Karamay of Xinjiang, West China. Infect. Dis. Poverty 2013, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Yuan, D.; Qin, H.; Zhang, J.; Liao, L.; Chen, Q.; Chen, D.; Chen, J. Phylogenetic analysis of HSP70 and cyt b gene sequences for Chinese Leishmania isolates and ultrastructural characteristics of Chinese Leishmania sp. Parasitol. Res. 2017, 116, 693–702. [Google Scholar] [CrossRef]
- Mukherjee, I.; Chakraborty, A.; Chakrabarti, S. Identification of internalin-A-like virulent proteins in Leishmania donovani. Parasites Vectors 2016, 9, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laffitte, M.N.; Leprohon, P.; Papadopoulou, B.; Ouellette, M. Plasticity of the Leishmania genome leading to gene copy number variations and drug resistance. F1000Research 2016, 5, 2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Prada, C.; Sharma, M.; Plourde, M.; Bresson, E.; Roy, G.; Leprohon, P.; Ouellette, M. High-throughput Cos-Seq screen with intracellular Leishmania infantum for the discovery of novel drug-resistance mechanisms. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, B.; Gazanion, E.; Mariac, C.; Du Manoir, M.; Sollelis, L.; Lopez-Rubio, J.J.; Sterkers, Y.; Bañuls, A.L. A single amino acid substitution (H451Y) in Leishmania calcium-dependent kinase SCAMK confers high tolerance and resistance to antimony. J. Antimicrob. Chemother. 2019, 74, 3231–3239. [Google Scholar] [CrossRef]
- Zheng, Z.; Chen, J.; Ma, G.; Satoskar, A.R.; Li, J. Integrative genomic, proteomic and phenotypic studies of Leishmania donovani strains revealed genetic features associated with virulence and antimony-resistance. Parasites Vectors 2020, 13, 510. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leishmania Species | Isolate | Country | Host | Abbreviation in This Study |
|---|---|---|---|---|
| Leishmania donovani | MHOM/IN/80/DD8 | India | Homo | L_DD8 |
| Leishmania donovania | MHOM/CN/80/801 | Kashgar, Xinjiang, China | Homo | L_801 |
| Leishmania donovani | MHOM/CN/90/9044 | Shandong, China | Homo | L_9044 |
| Leishmania infantuma | MHOM/CN/94/KXG-Liu | Karamay, Xinjiang, China | Homo | L_Liu |
| Leishmania donovanib | MHOM/CN/2016/SCHCZ | Sichuan, China | Homo | L_HCZ |
| GenBank Assembly Accession | Leishmania Species | Strain | Country | Host | Date |
|---|---|---|---|---|---|
| GCA_003730215.1 | Leishmania donovani | FDAARGOS_361 | Sudan | Homo | 2018/11/15 |
| GCA_000227135.2 | Leishmania donovani | BPK282A1 | Nepal | Homo | 2011/12/13 |
| GCA_000470725.1 | Leishmania donovani | BHU 1220 | India | Homo | 2013/09/30 |
| GCA_003719575.1 | Leishmania donovani | LdCL | Canada | Homo | 2018/11/09 |
| GCA_001989955.1 | Leishmania donovani | MHOM/IN/1983/AG83 | India | Homo | 2017/02/07 |
| GCA_001989975.1 | Leishmania donovani | MHOM/IN/1983/AG83 | India | Homo | 2017/02/07 |
| GCA_000002875.2 | Leishmania infantum | JPCM5 (MCAN/ES/98/LLM-877) | Spain | Canis | 2011/11/08 |
| GCA_900500625.1 | Leishmania infantum | JPCM5 | Spain | Canis | 2018/08/08 |
| GCA_003671315.1 | Leishmania infantum | HUUFS14 | Unknown | Homo | 2018/10/22 |
| GCA_003020905.1 | Leishmania infantum | TR01 | Turkey | Homo | 2018/03/27 |
| GCA_002243465.1 | Leishmania donovani | pasteur | France | Homo | 2017/08/08 |
| GCA_003730175.1 | Leishmania donovani | FDAARGOS_360 | Sudan | Homo | 2018/11/15 |
| GCA_000002725.2 | Leishmania major | Friedlin (MHOM/IL/81/Friedlin) | Israel | Homo | 2011/02/14 |
| GCA_000250755.2 | Leishmania major | SD 75.1 | Unknown | Unknown | 2012/03/07 |
| GCA_000331345.1 | Leishmania major | LV39c5 | Unknown | Unknown | 2013/01/11 |
| GCA_000441995.1 | Leishmania turanica | LEM423 (MMEL/SU/79/MEL) | Soviet union | Meles | 2013/07/29 |
| GCA_000443025.1 | Leishmania gerbilli | LEM452 (MRHO/CN/60/GERBILLI) | China | Rhombomys | 2013/08/25 |
| GCA_000410695.2 | Leishmania arabica | LEM1108 (MPSA/SA/83/JISH220) | Saudi Arabia | Psammomys | 2016/09/16 |
| GCA_000410715.1 | Leishmania tropica | L590 (MHOM/IL/1990/P283) | Israel | Homo | 2013/06/12 |
| GCA_003352575.1 | Leishmania tropica | MHOM/LB/2015/IK | Lebanon | Homo | 2018/08/01 |
| GCA_003067545.1 | Leishmania tropica | MHOM/LB/2017/IK | Lebanon | Homo | 2018/04/23 |
| GCA_000444285.2 | Leishmania aethiopica | L147 (MHOM/ET/1972/L100) | Ethiopia | Homo | 2016/09/30 |
| GCA_003992445.1 | Leishmania aethiopica | 209-622 | Ethiopia | Homo | 2019/01/04 |
| GCA_000438535.1 | Leishmania amazonensis | MHOM/BR/71973/M2269 | Brazil | Homo | 2013/07/18 |
| GCA_005317125.1 | Leishmania amazonensis | UA301 | Colombia | Homo | 2019/05/14 |
| GCA_003992505.1 | Leishmania amazonensis | 210-660 | French Guiana | Homo | 2019/01/04 |
| GCA_000234665.4 | Leishmania mexicana | MHOM/GT/2001/U1103 | Guatemala | Homo | 2011/11/08 |
| GCA_003992435.1 | Leishmania mexicana | 215-49 | USA | Homo | 2019/01/04 |
| Sample | Map Reads | Map Rate (%) | Average Depth (%) | Coverage 1× (%) | Coverage 4× (%) | Coverage 10× (%) |
|---|---|---|---|---|---|---|
| L_DD8 | 10,781,472 | 93.23 | 38.46 | 96.15 | 95.72 | 94.66 |
| L_801 | 9,059,366 | 93.50 | 32.53 | 96.10 | 95.51 | 93.70 |
| L_9044 | 11,278,920 | 90.41 | 38.05 | 96.09 | 95.68 | 94.79 |
| L_Liu | 11,870,398 | 93.93 | 42.81 | 96.09 | 95.67 | 94.79 |
| L_HCZ | 7,844,882 | 90.01 | 30.12 | 96.00 | 95.32 | 90.54 |
| Sample | Exonic Mutation | Aneuploidy Mutation | Aneuploidy Mutation Rate (%) |
|---|---|---|---|
| L. DD8 | 883 | 773 | 87.54 |
| L. 801 | 1022 | 918 | 89.28 |
| L. 9044 | 37,796 | 1648 | 4.36 |
| L. Liu | 38,078 | 2054 | 5.39 |
| L. HCZ | 49,352 | 1322 | 2.68 |
| Mutated Gene | Number of Mutations | ||||
|---|---|---|---|---|---|
| L_DD8 | L_801 | L_9044 | L_Liu | L_HCZ | |
| protein kinase | 5 | 4 | 971 | 980 | 1196 |
| dynein heavy chain | 1 | 6 | 353 | 355 | 416 |
| ATP-binding cassette protein subfamily A | 60 | 54 | 110 | 112 | 344 |
| kinesin | 6 | 6 | 226 | 231 | 312 |
| uncharacterized protein | 69 | 83 | 352 | 345 | 308 |
| calpain-like cysteine peptidase | 0 | 14 | 205 | 193 | 173 |
| amastin-like surface protein | 69 | 67 | 132 | 125 | 136 |
| ubiquitin hydrolase | 0 | 0 | 111 | 115 | 132 |
| phosphoglycan beta 1 | 11 | 9 | 94 | 92 | 100 |
| serine/threonine-protein kinase | 1 | 1 | 99 | 100 | 96 |
| N-acyl-L-amino acid amidohydrolase | 0 | 0 | 7 | 7 | 87 |
| RNA binding protein | 0 | 2 | 51 | 51 | 84 |
| folate/biopterin transporter | 13 | 11 | 80 | 90 | 70 |
| phosphatidylinositol 3-kinase-like protein | 0 | 1 | 40 | 40 | 69 |
| protein-like multidrug resistant | 0 | 0 | 16 | 16 | 65 |
| amastin-like protein | 7 | 4 | 43 | 39 | 56 |
| kinetoplast-associated protein-like protein | 0 | 0 | 57 | 56 | 55 |
| receptor-type adenylate cyclase a | 26 | 29 | 30 | 35 | 53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Z.; He, J.; Luo, T.; Zhang, J.; Zhou, Q.; Yin, S.; Chen, D.; Luo, J.; Chen, J.; Li, J. Mutation Characteristics and Phylogenetic Analysis of Five Leishmania Clinical Isolates. Animals 2022, 12, 321. https://doi.org/10.3390/ani12030321
Zheng Z, He J, Luo T, Zhang J, Zhou Q, Yin S, Chen D, Luo J, Chen J, Li J. Mutation Characteristics and Phylogenetic Analysis of Five Leishmania Clinical Isolates. Animals. 2022; 12(3):321. https://doi.org/10.3390/ani12030321
Chicago/Turabian StyleZheng, Zhiwan, Jinlei He, Tao Luo, Jianhui Zhang, Qi Zhou, Shuangshuang Yin, Dali Chen, Jie Luo, Jianping Chen, and Jiao Li. 2022. "Mutation Characteristics and Phylogenetic Analysis of Five Leishmania Clinical Isolates" Animals 12, no. 3: 321. https://doi.org/10.3390/ani12030321