
Effect of Different Dietary Regimes on the Gut Microbiota and Fecal Metabolites of Père David’s Deer

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Diet Survey and Analysis
2.2. Fecal Sample Collection
2.3. Microscopic Examination and Identification
2.4. 16. S rRNA Microbial Community Analysis
2.5. Sequence Processing and Analysis
2.6. UPLC-MS/MS Analysis
2.7. Metabolomic Analysis
2.8. Statistical Analysis
3. Results
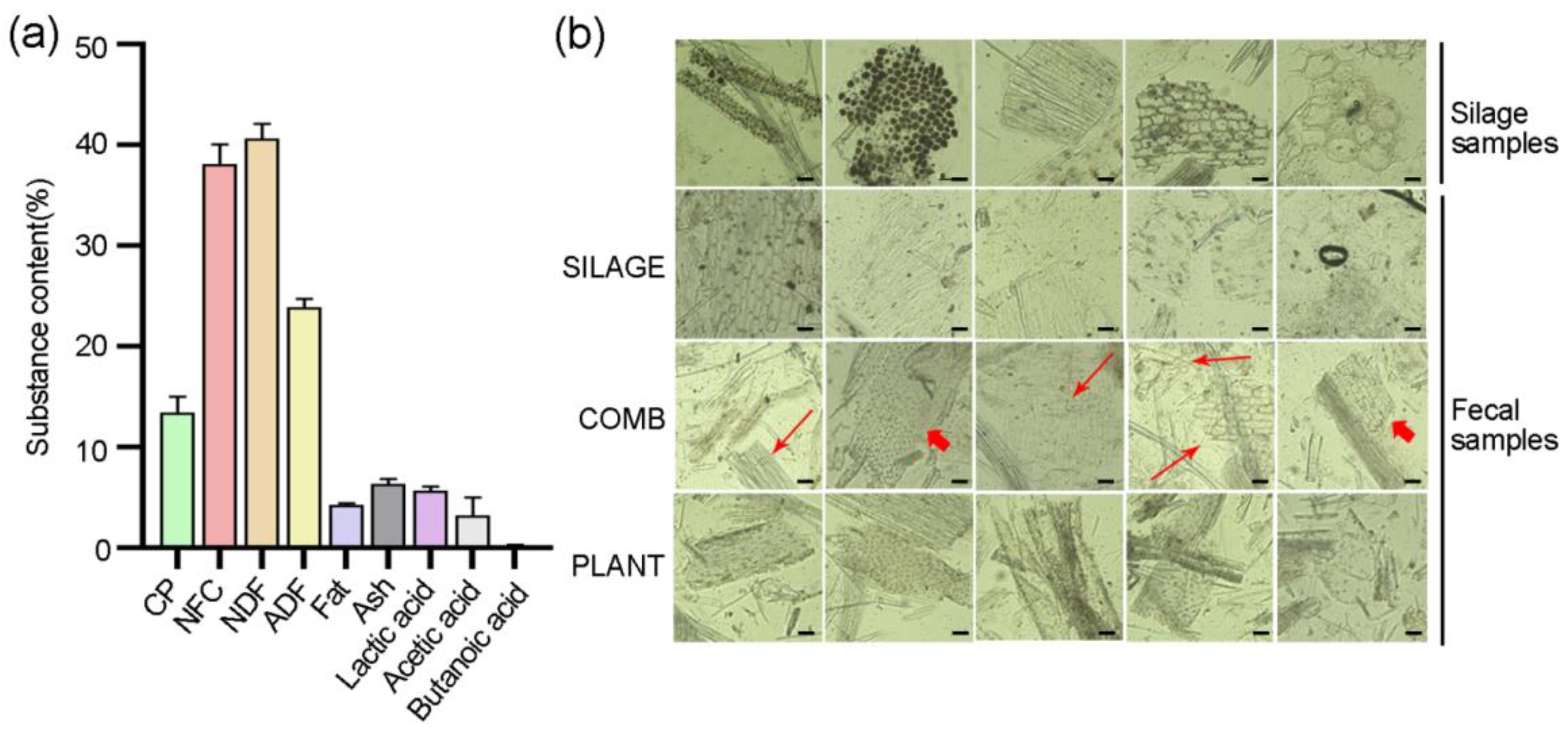
3.1. Diet Identification from Fecal Samples
3.2. Effect of Different Diets on the Diversity of the Gut Microbiota
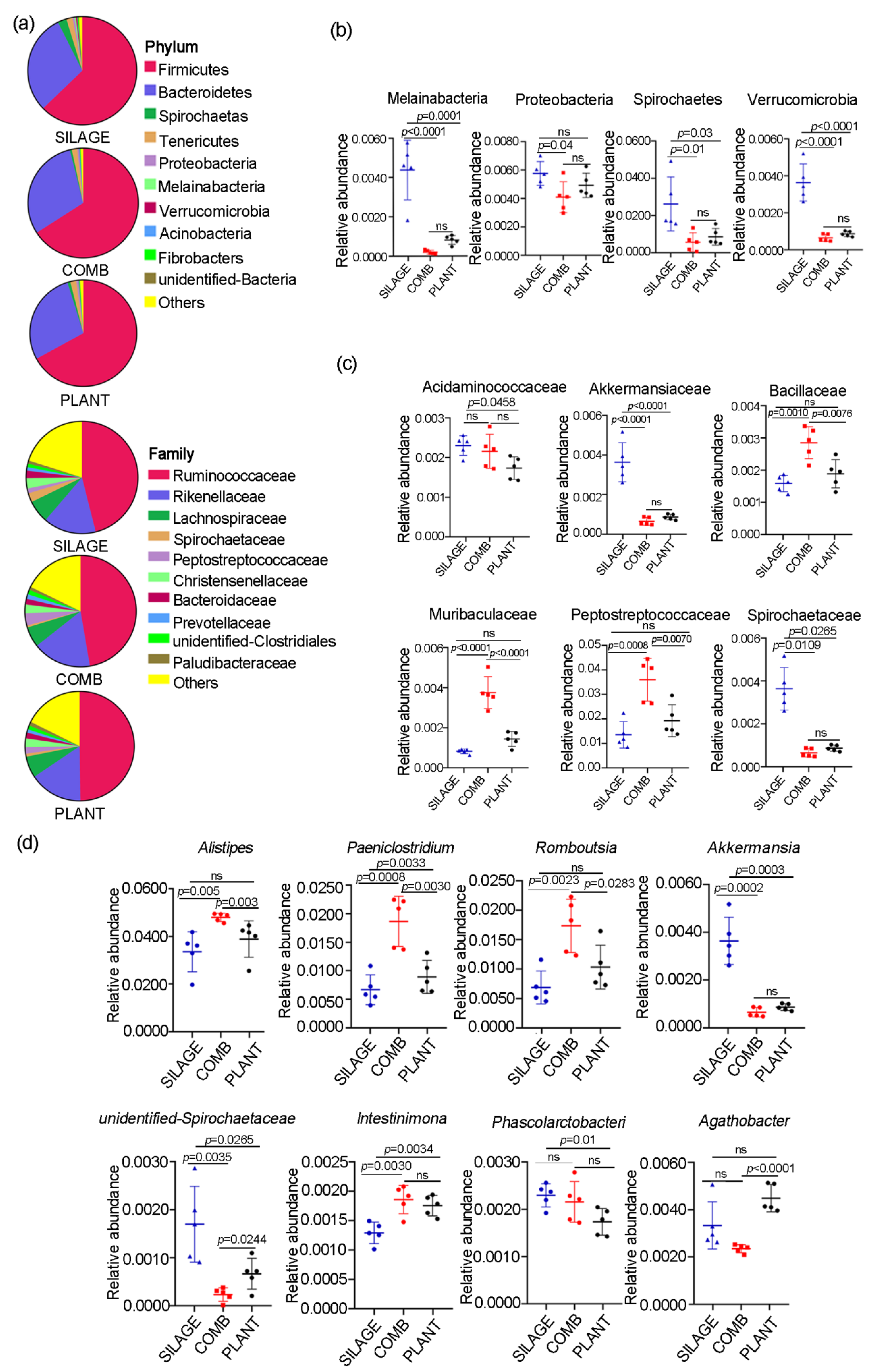
3.3. Gut Microbiota Composition of Père David’s Deer Was Altered with the Different Dietary Regimes
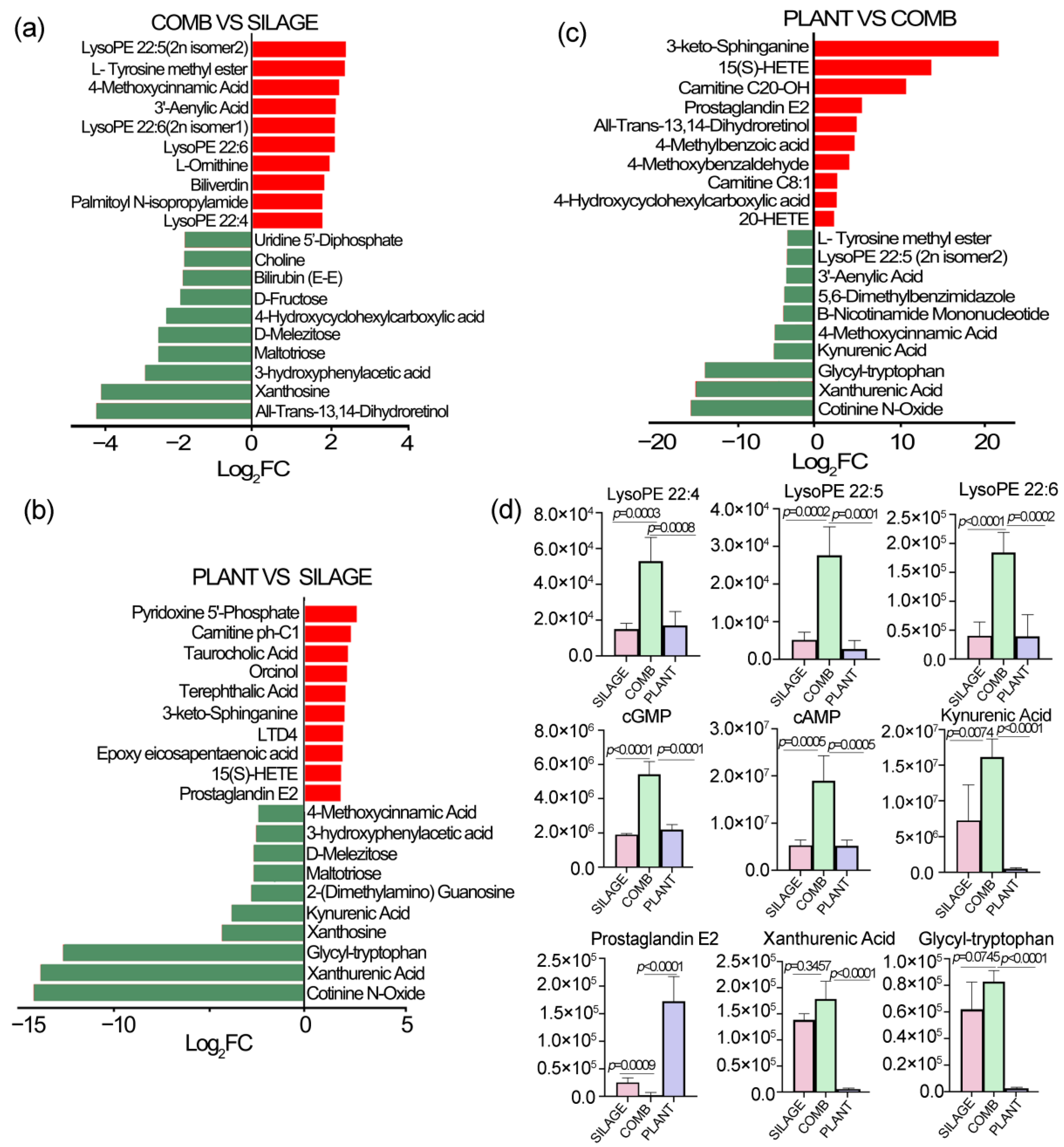
3.4. Fecal Metabolite Profiles Were Different under the Different Dietary Regimes
3.5. Differences of Fecal Metabolites under the Different Dietary Regimes
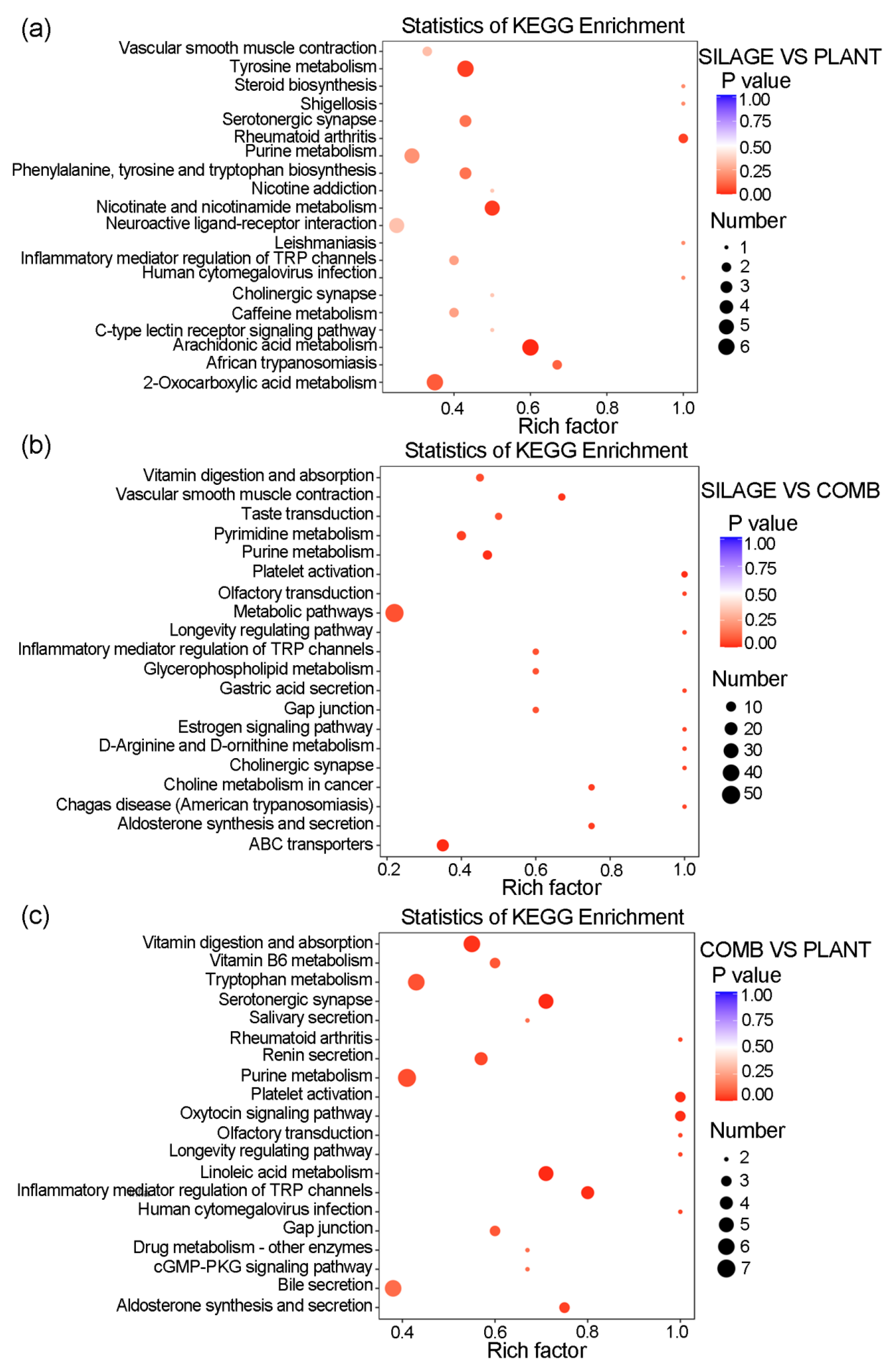
3.6. Several Metabolic Pathways Were Different under the Different Dietary Regimes
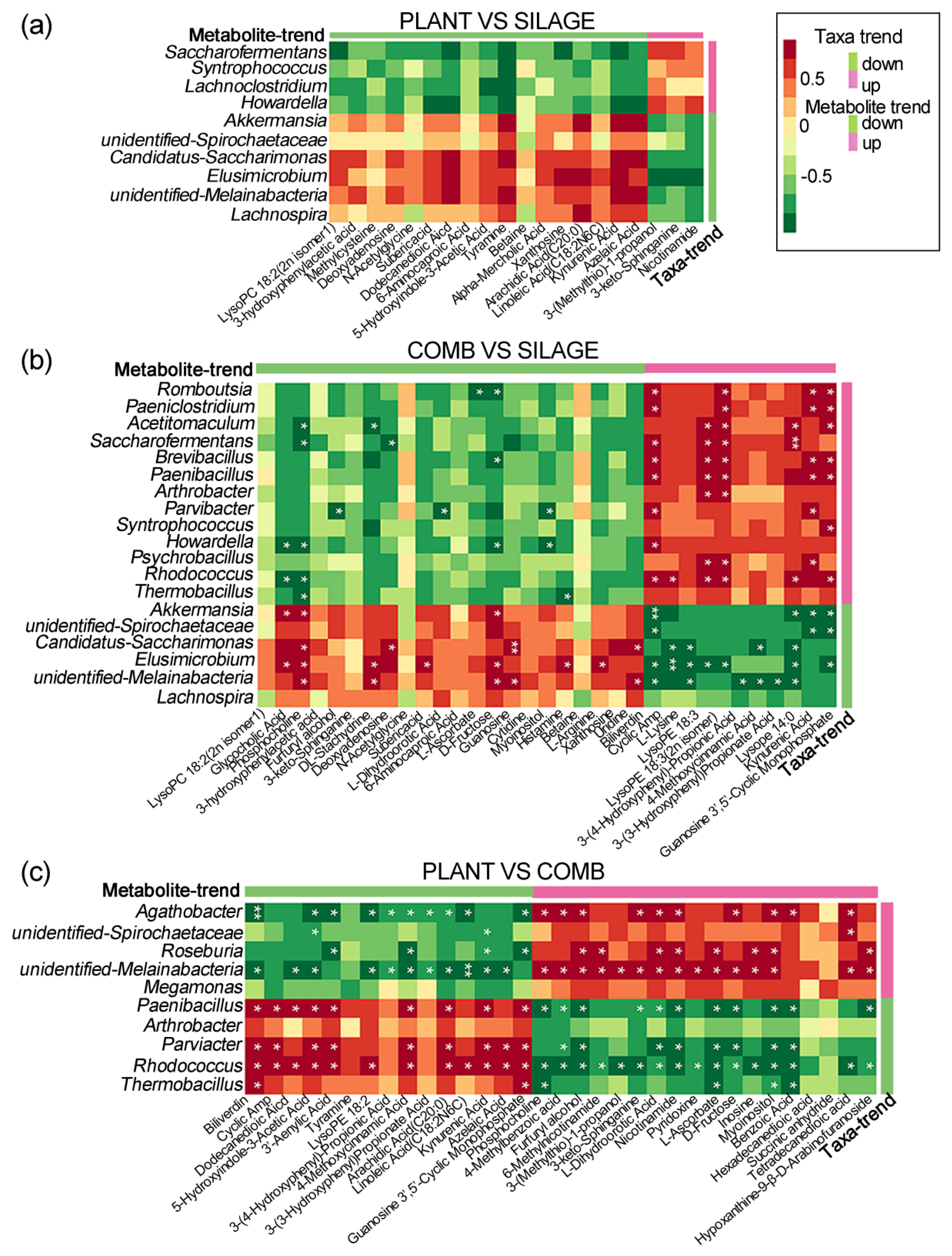
3.7. Different Gut Microbiota Related to the Metabolic Phenotype under the Different Dietary Regimes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, B.; Ji, Y.; Wang, J.; Pei, Q. The Annual Habitat Selection of Released Père David’s Deer in Dafeng Milu National Nature Reserve. Acta Ecol. Sin. 2011, 31, 225–232. [Google Scholar] [CrossRef]
- Jiang, Z.; Yu, C.; Feng, Z.; Zhang, L.; Xia, J.; Ding, Y.; Lindsay, N. Reintroduction and Recovery of Père David’s Deer in China. Wildl. Soc. 2000, 28, 681–687. [Google Scholar]
- Zhang, S.; Li, C.; Li, Y.; Chen, Q.; Hu, D.; Cheng, Z.; Wang, X.; Shan, Y.; Bai, J.; Liu, G. Genetic Differentiation of Reintroduced Père David’s Deer (Elaphurus davidianus) Based on Population Genomics Analysis. Front. Genet. 2021, 12, 705337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bai, J.; Zhu, A.; Chen, R.; Xue, D.; Zhong, Z.; Cheng, Z. Reversing Extinction in China’s Père David’s Deer. Science 2021, 371, 685. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.H.; Liu, H.Y.; Liu, B.; Yuan, B.D.; Lu, C.H. Analysis of the Gut Microbiome of Wild and Captive Père David’s Deer. Front. Microbiol. 2019, 10, 2331. [Google Scholar] [CrossRef]
- Hicks, A.L.; Lee, K.J.; Couto-Rodriguez, M.; Patel, J.; Sinha, R.; Guo, C.; Olson, S.H.; Seimon, A.; Seimon, T.A.; Ondzie, A.U.; et al. Gut microbiomes of Wild Great Apes Fluctuate Seasonally in Response to Diet. Nat. Commun. 2018, 9, 1786. [Google Scholar] [CrossRef] [Green Version]
- Stedman, A.; van Vliet, A.H.M.; Chambers, M.A.; Gutierrez-Merino, J. Gut Commensal Bacteria Show Beneficial Properties as Wildlife Probiotics. Ann. N. Y. Acad. Sci. 2020, 1467, 112–132. [Google Scholar] [CrossRef] [Green Version]
- Morrison, K.E.; Jašarević, E.; Howard, C.D.; Bale, T.L. It’s the Fiber, Not the Fat: Significant Effects of Dietary Challenge on the Gut Microbiome. Microbiome 2020, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; Qi, J.; Dobbins, M.T.; Liang, X.; Wang, J.; Chen, S.; Ma, J.; Jiang, G. Comparative Analysis of Microbial Community Structure and Function in the Gut of Wild and Captive Amur Tiger. Front. Microbiol. 2020, 11, 1665. [Google Scholar] [CrossRef]
- Liu, K.; Wang, L.; Yan, T.; Wang, Z.; Xue, B.; Peng, Q. Relationship Between the Structure and Composition of Rumen Microorganisms and the Digestibility of Neutral Detergent Fibre in Goats. Asian-Australas J. Anim. Sci. 2019, 32, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shi, M.; Fan, M.; Xu, S.; Li, Y.; Zhang, T.; Cha, M.; Liu, Y.; Guo, X.; Chen, Q.; et al. Comparative Analysis of Gut Microbiota Changes in Père David’s Deer Populations in Beijing Milu Park and Shishou, Hubei Province in China. Front. Microbiol. 2018, 9, 1258. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ding, J.; Yang, Z.; Chen, H.; Yao, R.; Dai, Q.; Ding, Y.; Zhu, L. Père David’s Deer Gut Microbiome Changes across Captive and Translocated Populations: Implications for Conservation. Evol. Appl. 2019, 12, 622–635. [Google Scholar] [CrossRef]
- Takewaki, D.; Suda, W.; Sato, W.; Takayasu, L.; Kumar, N.; Kimura, K.; Kaga, N.; Mizuno, T.; Miyake, S.; Hattori, M.; et al. Alterations of the Gut Ecological and Functional Microenvironment in Different Stages of Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2020, 36, 22402–22412. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Jin, G.; Wang, G.; Liu, T.; Liu, X.; Wang, B.; Cao, H. Current Sampling Methods for Gut Microbiota: A Call for More Precise Devices. Front. Cell Infect. Microbiol. 2020, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zhai, J.; Zhang, L.; Liu, D.; Ma, Y.; Rong, K.; Xu, Y.C.; Ma, J. Variations in Gut Microbiota and Fecal Metabolic Phenotype Associated with Fenbendazole and Ivermectin Tablets by 16S rRNA Gene Sequencing and LC/MS-based Metabolomics in Amur Tiger. Biochem. Biophys. Res. Commun. 2018, 499, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Cho, H.; Lee, W.Y. Distinct Gut Microbiotas between Southern Elephant Seals and Weddell Seals of Antarctica. J. Microbiol. 2020, 58, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Michonneau, D.; Latis, E.; Curis, E.; Dubouchet, L.; Ramamoorthy, S.; Ingram, B.; de Latour, R.P.; Robin, M.; de Fontbrune, F.S.; Chevret, S.; et al. Metabolomics Analysis of Human Acute Graft-versus-host Disease Reveals Changes in Host and Microbiota-derived Metabolites. Nat. Commun. 2019, 10, 5695. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.P.; Zhang, S.M.; Liu, Y.J.; Bai, J.D. Analysis of Nutrient Composition of Elk’s Favorite Plants in Different Habitats. Heilongjiang Anim. Sci. Vet. Med. 2017, 9, 252–255. (In Chinese) [Google Scholar]
- Hou, L.B.; Ding, J.J.; Ding, Y.H.; Ren, Y.J. Relationship between the Climate Change and Survival of Milu. J. Beijing For. Univ. 2011, A2, 11–15. (In Chinese) [Google Scholar]
- Hugerth, L.W.; Wefer, H.A.; Lundin, S.; Jakobsson, H.E.; Lindberg, M.; Rodin, S.; Engstrand, L.; Andersson, A.F. DegePrime, a Program for Degenerate Primer Design for Broad-taxonomic-range PCR in Microbial Ecology Studies. Appl. Environ. Microbiol. 2014, 80, 5116–5123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA Sequence Formation and Detection in Sanger and 454-pyrosequenced PCR Amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-discovery Approach for Sample Matching of a Nerve-agent Precursor Using Liquid Chromatography-mass Spectrometry, XCMS, and Chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.M.; Rogers, T.L.; Carlini, A.R.; Brown, M.V. Diet and Phylogeny Shape the Gut Microbiota of Antarctic Seals: A Comparison of Wild and Captive Animals. Environ. Microbiol. 2013, 15, 1132–1145. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.M.; Nguyen, B.N.; Neumann, L.M.; Miller, M.; Buss, P.; Daniels, S.; Ahn, M.J.; Crandall, K.A.; Pukazhenthi, B. Gut Microbiome Differences between Wild and Captive Black Rhinoceros—Implications for Rhino Health. Sci. Rep. 2019, 9, 7570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Chi, X.; Qin, W.; Wang, L.; Song, P.; Cai, Z.; Zhang, J.; Zhang, T. Comparison of the Gut Microbiota Composition between the Wild and Captive Tibetan Wild Ass (Equus kiang). J. Appl. Microbiol. 2019, 126, 1869–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, V.J.; Song, S.J.; Delsuc, F.; Prest, T.L.; Oliverio, A.M.; Korpita, T.M.; Alexiev, A.; Amato, K.R.; Metcalf, J.L.; Kowalewski, M.; et al. The Effects of Captivity on the Mammalian Gut Microbiome. Integr. Comp. Biol. 2017, 57, 690–704. [Google Scholar] [CrossRef] [Green Version]
- Clayton, J.B.; Vangay, P.; Huang, H.; Ward, T.; Hillmann, B.M.; Al-Ghalith, G.A.; Travis, D.A.; Long, H.T.; Tuan, B.V.; Minh, V.V.; et al. Captivity Humanizes the Primate Microbiome. Proc. Natl. Acad. Sci. USA 2016, 113, 10376–10381. [Google Scholar] [CrossRef] [Green Version]
- Manor, O.; Dai, C.L.; Kornilov, S.A.; Smith, B.; Price, N.D.; Lovejoy, J.C.; Gibbons, S.M.; Magis, A.T. Health and Disease Markers Correlate with Gut Microbiome Composition across Thousands of People. Nat. Commun. 2020, 11, 5206. [Google Scholar] [CrossRef]
- Hu, Q.; Wu, K.; Pan, W.; Zeng, Y.; Hu, K.; Chen, D.; Huang, X.; Zhang, Q. Intestinal Flora Alterations in Patients with Early Chronic Kidney Disease: A Case-Control Study among the Han Population in Southwestern China. J. Int. Med. Res. 2020, 48, 300060520926033. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Uchiyama, K.; Takagi, T. A Next-Generation Beneficial Microbe: Akkermansia Muciniphila. J. Clin. Biochem. Nutr. 2018, 63, 33–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchione, I.G.; Lopetuso, L.R.; Ianiro, G.; Napoli, M.; Gibiino, G.; Rizzatti, G.; Petito, V.; Gasbarrini, A.; Scaldaferri, F. Akkermansia Muciniphila: Key Player in Metabolic and Gastrointestinal Disorders. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8075–8083. [Google Scholar]
- Lübeck, M.; Lübeck, P.S. Application of Lactic Acid Bacteria in Green Biorefineries. FEMS Microbiol. Lett. 2019, 366, fnz024. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Xiang, Y.; Zhang, J.; Luo, N.; Liang, M.; Gong, L.; Yu, J.; Cui, Q.; Sepulcre, J.; Chen, H. A Reachable Probability Approach for the Analysis of Spatio-Temporal Dynamics in the Human Functional Network. Neuroimage 2021, 243, 118497. [Google Scholar] [CrossRef]
- Wang, H.; Xu, R.; Zhang, H.; Su, Y.; Zhu, W. Swine Gut Microbiota and Its Interaction with Host Nutrient Metabolism. Anim. Nutr. 2020, 6, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Wang, P.; Fu, T.; Lu, M.; Cai, Y.; Chen, X.; Cheng, L. A Comprehensive Review for Gut Microbes: Technologies, Iinterventions, Metabolites and Diseases. Brief Funct. Genom. 2021, 20, 42–60. [Google Scholar] [CrossRef]
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and Biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Hasan, R.A.; Koh, A.Y.; Zia, A. The Gut Microbiome and Thromboembolism. Thromb. Res. 2020, 189, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Alhouayek, M.; Ameraoui, H.; Muccioli, G.G. Bioactive Lipids in Iinflammatory Bowel Diseases—From Pathophysiological Alterations to Therapeutic Opportunities. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158854. [Google Scholar] [CrossRef]
- Zou, D.; Pei, J.; Lan, J.; Sang, H.; Chen, H.; Yuan, H.; Wu, D.; Zhang, Y.; Wang, Y.; Wang, D.; et al. A SNP of Bacterial Blc Disturbs Gut Lysophospholipid Homeostasis and Induces Inflammation through Epithelial Barrier Disruption. EBioMedicine 2020, 52, 102652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalvon-Demersay, T.; Luise, D.; Le Floc’h, N.; Tesseraud, S.; Lambert, W.; Bosi, P.; Trevisi, P.; Beaumont, M.; Corrent, E. Functional, Amino Acids in Pigs and Chickens: Implication for Gut Health. Front. Vet. Sci. 2021, 8, 663727. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Taleb, S. Tryptophan Dietary Impacts Gut Barrier and Metabolic Diseases. Front. Immunol. 2019, 10, 2113. [Google Scholar] [CrossRef] [PubMed]
- Rudzki, L.; Stone, T.W.; Maes, M.; Misiak, B.; Samochowiec, J.; Szulc, A. Gut Microbiota-Derived Vitamins-Underrated Powers of a Multipotent Ally in Psychiatric Health and Disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 107, 110240. [Google Scholar] [CrossRef] [PubMed]
- Leng, X.; Jiang, H. Effects of Arachidonic Acid and Its Major Prostaglandin Derivatives on Bovine Myoblast Proliferation, Differentiation, and Fusion. J. Domest. Anim. Endocrinol. 2019, 67, 28–36. [Google Scholar] [CrossRef]
- Monmai, C.; Jang, A.Y.; Kim, J.E.; Lee, S.M.; You, S.; Kang, S.; Park, W.J. Immunomodulatory Activities of Body Wall Fatty Acids Extracted from Halocynthia aurantium on RAW264.7 Cells. J. Microbiol. Biotechnol. 2020, 30, 1927–1936. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhen, J.; Ren, Y.; Zhang, H.; Yuan, X.; Wang, L.; Shen, H.; Liu, P.; Chen, Y. Effect of Different Dietary Regimes on the Gut Microbiota and Fecal Metabolites of Père David’s Deer. Animals 2022, 12, 584. https://doi.org/10.3390/ani12050584
Zhen J, Ren Y, Zhang H, Yuan X, Wang L, Shen H, Liu P, Chen Y. Effect of Different Dietary Regimes on the Gut Microbiota and Fecal Metabolites of Père David’s Deer. Animals. 2022; 12(5):584. https://doi.org/10.3390/ani12050584
Chicago/Turabian StyleZhen, Junai, Yijun Ren, Huidan Zhang, Xueli Yuan, Libo Wang, Hua Shen, Ping Liu, and Yuqing Chen. 2022. "Effect of Different Dietary Regimes on the Gut Microbiota and Fecal Metabolites of Père David’s Deer" Animals 12, no. 5: 584. https://doi.org/10.3390/ani12050584
APA StyleZhen, J., Ren, Y., Zhang, H., Yuan, X., Wang, L., Shen, H., Liu, P., & Chen, Y. (2022). Effect of Different Dietary Regimes on the Gut Microbiota and Fecal Metabolites of Père David’s Deer. Animals, 12(5), 584. https://doi.org/10.3390/ani12050584