
Differences in Faecal Microbiome Taxonomy, Diversity and Functional Potential in a Bovine Cohort Experimentally Challenged with Mycobacterium avium subsp. paratuberculosis (MAP)
 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. DNA Extractions and Library Preparations
2.3. Bioinformatics Analysis of Exposed vs. Control Animals across Time
2.4. Statistical Analysis Comparing Exposed to Controls and across Time, and Correlating Immunopathology with Taxonomic Output
3. Results
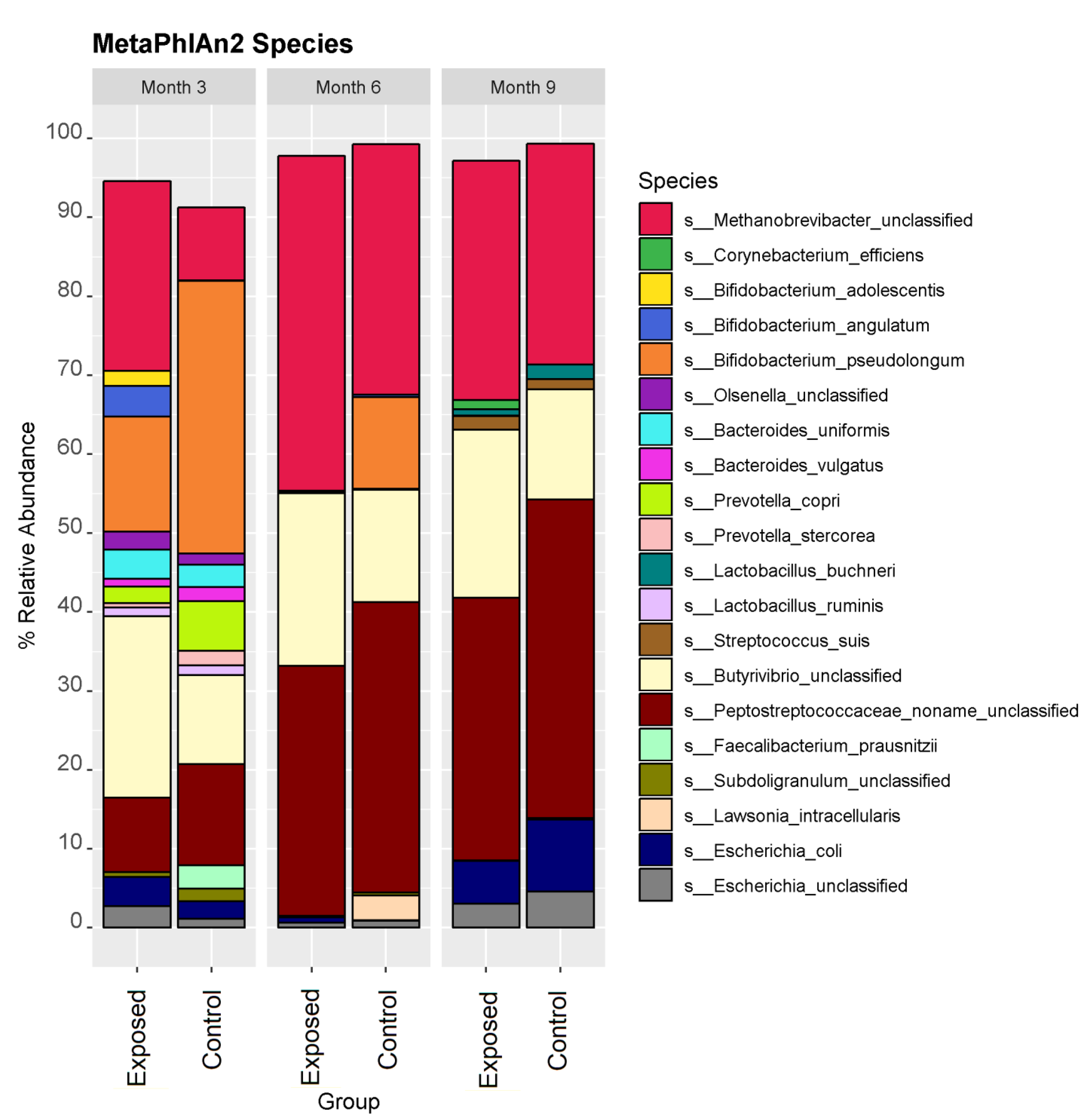
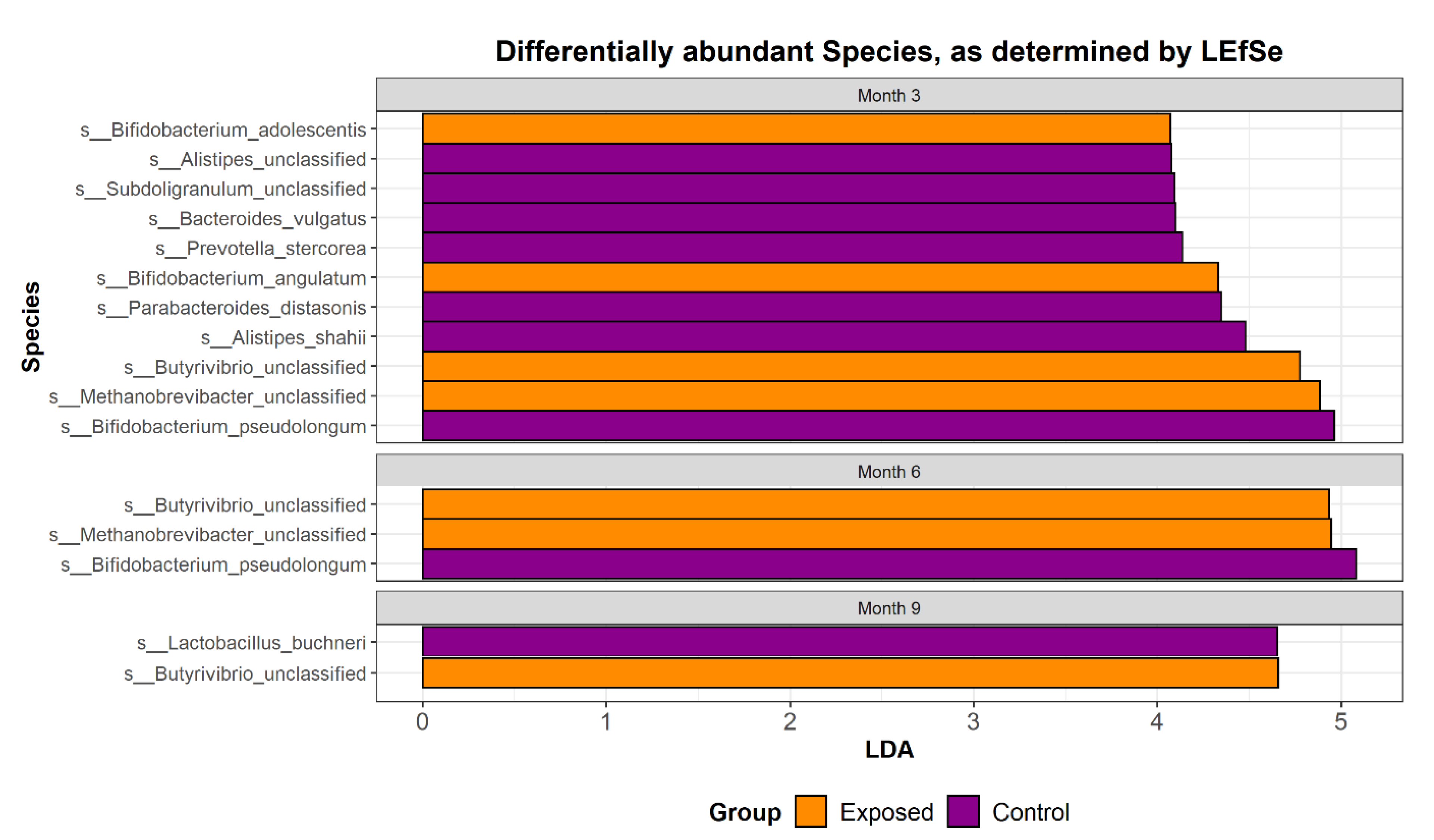
3.1. The Taxonomic Profile of Calves Exposed to MAP Differs from Controls
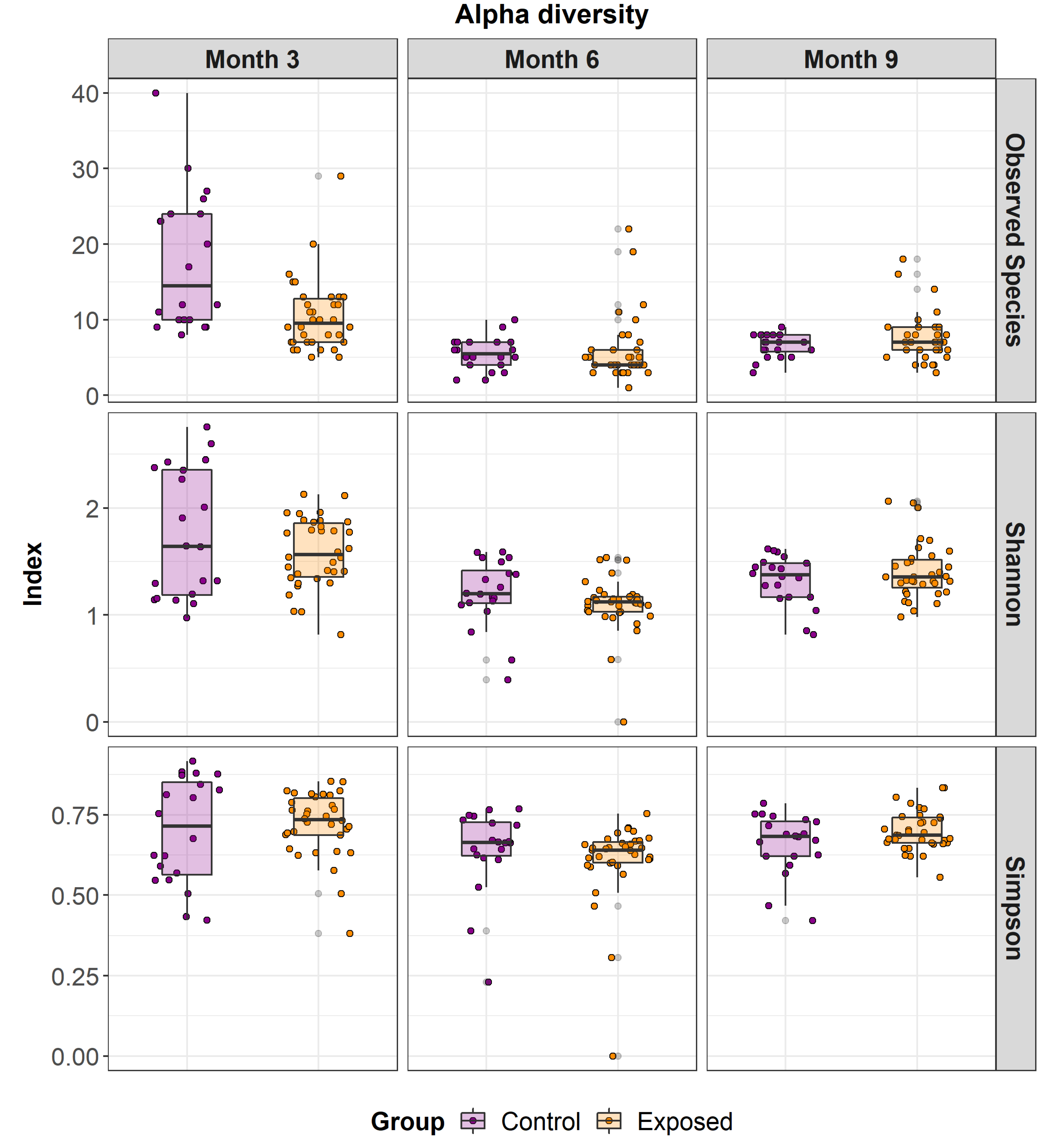
3.2. Differences in Faecal Microbiome Alpha and Beta Diversity between MAP Exposed and Control Animals
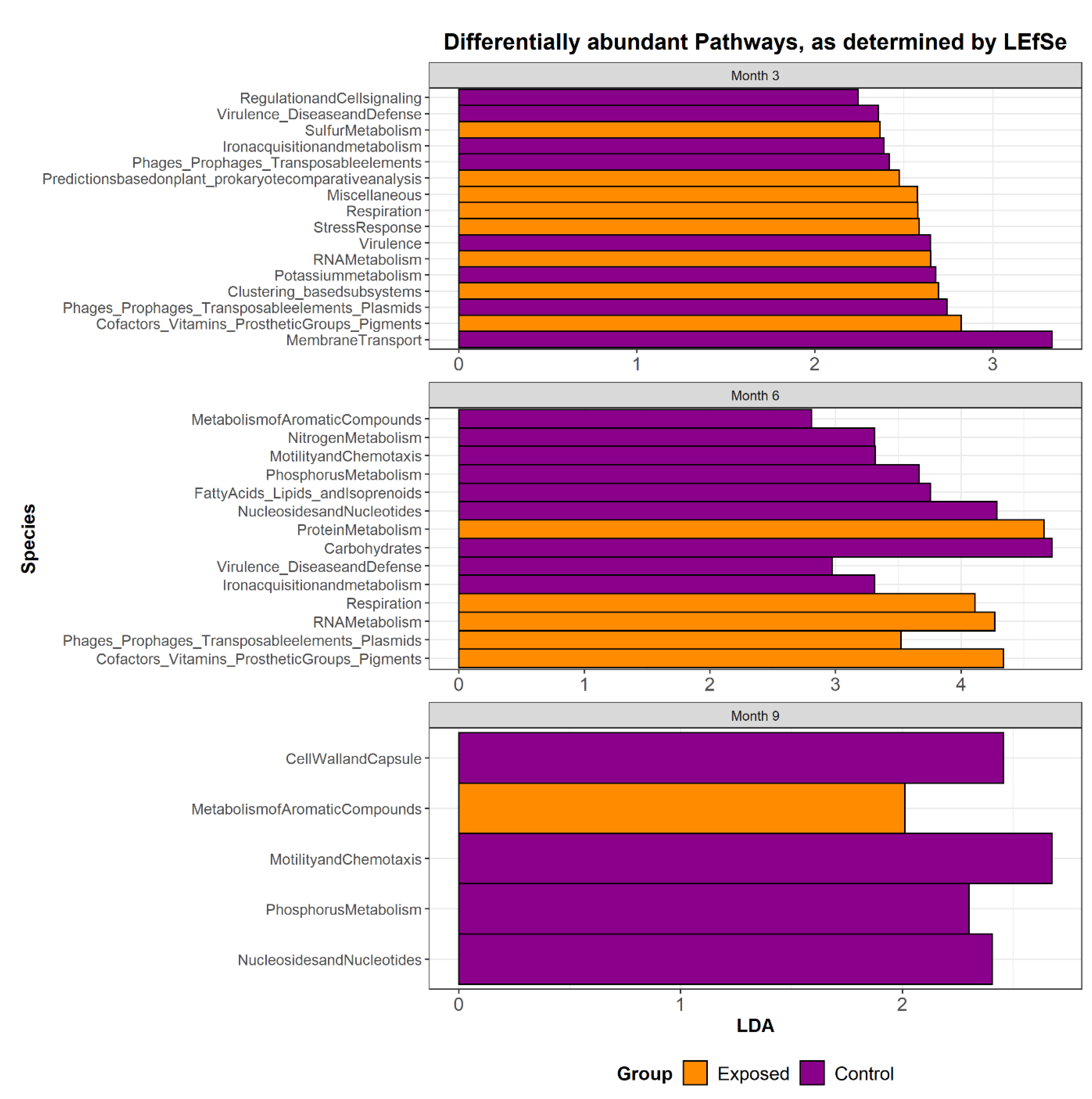
3.3. The Functional Potential of the Microbiome of Animals Exposed to MAP Differs from That of Controls
3.4. Correlations between the Gut Microbiome and Immunopathology Measurements Reveals Important Associations between the Microbiome and Host Immune Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DNA | Deoxyribonucleic acid |
| GIT | Gastrointestinal tract |
| IFN-ɣ | Interferon gamma |
| JD | Johne’s Disease |
| LDA | Linear discriminant analysis |
| MAP | Mycobacterium avium subsp. paratuberculosis |
| miRNA | microRNA |
| PERMANOVA | Permutational multivariate analysis of variance |
| qPCR | real-time PCR |
References
- Bate, A.M.; Jones, G.; Kleczkowski, A.; Naylor, R.; Timmis, J.; White, P.C.L.; Touza, J. Livestock Disease Management for Trading Across Different Regulatory Regimes. EcoHealth 2018, 15, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Pitta, D.W.; Pinchak, W.E.; Indugu, N.; Vecchiarelli, B.; Sinha, R.; Fulford, J.D. Metagenomic Analysis of the Rumen Microbiome of Steers with Wheat-Induced Frothy Bloat. Front. Microbiol. 2016, 7, 689. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Crispie, F.; O’Sullivan, O.; Finnegan, L.; Claesson, M.J.; Cotter, P.D. Species Clas-sifier Choice Is a Key Consideration When Analysing Low-Complexity Food Microbiome Data. Microbiome 2018, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Rosa, B.A.; Supali, T.; Gankpala, L.; Djuardi, Y.; Sartono, E.; Zhou, Y.; Fischer, K.; Martin, J.; Tyagi, R.; Bolay, F.K.; et al. Differential Human Gut Microbiome Assemblages during Soil-Transmitted Helminth Infections in Indonesia and Liberia. Microbiome 2018, 6, 33. [Google Scholar] [CrossRef]
- Patra, A.; Park, T.; Kim, M.; Yu, Z. Rumen Methanogens and Mitigation of Methane Emission by Anti-Methanogenic Compounds and Substances. J. Anim. Sci. Biotechnol. 2017, 8, 13. [Google Scholar] [CrossRef]
- Muñoz-Vargas, L.; Opiyo, S.O.; Digianantonio, R.; Williams, M.L.; Wijeratne, A.; Habing, G. Fecal Microbiome of Periparturient Dairy Cattle and Associations with the Onset of Salmonella Shedding. PLoS ONE 2018, 13, e0196171. [Google Scholar] [CrossRef]
- Li, R.W.; Wu, S.; Li, W.; Huang, Y.; Gasbarre, L.C. Metagenome Plasticity of the Bovine Abomasal Microbiota in Immune Animals in Response to Ostertagia Ostertagi Infection. PLoS ONE 2011, 6, e24417. [Google Scholar] [CrossRef]
- Naqid, I.A.; Owen, J.P.; Maddison, B.C.; Gardner, D.S.; Foster, N.; Tchórzewska, M.A.; La Ragione, R.M.; Gough, K.C. Prebiotic and Probiotic Agents Enhance Antibody-Based Immune Responses to Salmonella Typhimurium Infection in Pigs. Anim. Feed. Sci. Technol. 2015, 201, 57–65. [Google Scholar] [CrossRef]
- Argüello, H.; Estellé, J.; Leonard, F.C.; Crispie, F.; Cotter, P.D.; O’Sullivan, O.; Lynch, H.; Walia, K.; Duffy, G.; Lawlor, P.G.; et al. Influence of the Intestinal Microbiota on Colonization Resistance to Salmonella and the Shedding Pattern of Naturally Exposed Pigs. mSystems 2019, 4, e00021-19. [Google Scholar] [CrossRef]
- Heras, V.L.; Clooney, A.G.; Ryan, F.J.; Cabrera-Rubio, R.; Casey, P.G.; Hueston, C.M.; Pinheiro, J.; Rudkin, J.K.; Melgar, S.; Cotter, P.D.; et al. Short-Term Consumption of a High-Fat Diet In-creases Host Susceptibility to Listeria Monocytogenes Infection. Microbiome 2019, 7, 7. [Google Scholar] [CrossRef]
- Hryckowian, A.J.; Van Treuren, W.; Smits, S.A.; Davis, N.M.; Gardner, J.O.; Bouley, D.M.; Sonnenburg, J.L. Microbiota-Accessible Carbohydrates Suppress Clostridium Difficile Infection in a Murine Model. Nat. Microbiol. 2018, 3, 662–669. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Griebel, P.J.; Guan, L.L. The Gut Microbiome and Its Potential Role in the De-velopment and Function of Newborn Calf Gastrointestinal Tract. Front. Vet. Sci. 2015, 2, 36. [Google Scholar] [CrossRef]
- Ma, T.; Villot, C.; Renaud, D.; Skidmore, A.; Chevaux, E.; Steele, M.; Guan, L.L. Linking Perturba-tions to Temporal Changes in Diversity, Stability, and Compositions of Neonatal Calf Gut Microbiota: Prediction of Diarrhea. ISME J. 2020, 14, 2223–2235. [Google Scholar] [CrossRef]
- Kargar, S.; Roshan, M.; Ghoreishi, S.M.; Akhlaghi, A.; Kanani, M.; Abedi Shams-Abadi, A.R.; Ghaffari, M.H. Extended Colostrum Feeding for 2 Weeks Improves Growth Performance and Re-duces the Susceptibility to Diarrhea and Pneumonia in Neonatal Holstein Dairy Calves. J. Dairy Sci. 2020, 103, 8130–8142. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Griebel, P.J.; Guan, L.L. Taxonomic Identification of Commensal Bacteria Asso-ciated with the Mucosa and Digesta throughout the Gastrointestinal Tracts of Preweaned Calves. Appl. Environ. Microbiol. 2014, 80, 2021–2028. [Google Scholar] [CrossRef]
- Windsor, P.A.; Whittington, R.J. Evidence for Age Susceptibility of Cattle to Johne’s Disease. Vet. J. 2010, 184, 37–44. [Google Scholar] [CrossRef]
- Derakhshani, H.; De Buck, J.; Mortier, R.; Barkema, H.W.; Krause, D.O.; Khafipour, E. The Features of Fecal and Ileal Mucosa-Associated Microbiota in Dairy Calves during Early Infection with My-cobacterium Avium Subspecies Paratuberculosis. Front. Microbiol. 2016, 7, 426. [Google Scholar] [CrossRef]
- Kaevska, M.; Videnska, P.; Sedlar, K.; Bartejsova, I.; Kralova, A.; Slana, I. Faecal Bacterial Composition in Dairy Cows Shedding Mycobacterium Avium Subsp. Paratuberculosis in Faeces in Comparison with Nonshedding Cows. Can. J. Microbiol. 2016, 62, 538–541. [Google Scholar] [CrossRef]
- Fecteau, M.-E.; Pitta, D.W.; Vecchiarelli, B.; Indugu, N.; Kumar, S.; Gallagher, S.C.; Fyock, T.L.; Sweeney, R.W. Dysbiosis of the Fecal Microbiota in Cattle Infected with Mycobacterium Avium Subsp. Paratuberculosis. PLoS ONE 2016, 11, e0160353. [Google Scholar] [CrossRef]
- Fernández, B.; Colavecchia, S.B.; Ingratta, G.G.; Jolly, A.; Stempler, A.; Fortuny, M.L.; Paolicchi, F.A.; Mundo, S.L. Early IgG2 in Calves Experimentally Infected with Mycobacterium Avium Subsp. Paratuberculosis. Vet. Immunol. Immunopathol. 2019, 213, 109886. [Google Scholar] [CrossRef] [PubMed]
- Begg, D.J.; Plain, K.M.; de Silva, K.; Gurung, R.; Gunn, A.; Purdie, A.C.; Whittington, R.J. Immunopathological Changes and Apparent Recovery from Infection Revealed in Cattle in an Experimental Model of Johne’s Disease Using a Lyophilised Culture of Mycobacterium Avium Subspecies Paratuberculosis. Vet. Microbiol. 2018, 219, 53–62. [Google Scholar] [CrossRef]
- Payne, J.M.; Rankin, J.D. The Pathogenesis of Experimental Johne’s Disease in Calves. Res. Vet. Sci. 1961, 2, 167–176. [Google Scholar] [CrossRef]
- Gilmour, N.J.L.; Nisbet, D.I.; Brotherston, J.G. Experimental Oral Infection of Calves with Mycobacterium Johnei. J. Comp. Pathol. 1965, 75, 281–286. [Google Scholar] [CrossRef]
- Farrell, D.; Shaughnessy, R.G.; Britton, L.; MacHugh, D.E.; Markey, B.; Gordon, S.V. The Identification of Circulating MiRNA in Bovine Serum and Their Potential as Novel Biomarkers of Early Mycobacterium Avium Subsp Paratuberculosis Infection. PLoS ONE 2015, 10, e0134310. [Google Scholar] [CrossRef] [PubMed]
- Britton, L.E. The Immunopathology of Mycobacterium Avium Subspecies Paratuberculosis (MAP) Infection in Cattle as a Basis for Novel Diagnostics. Ph.D. Thesis, University College Dublin, Dublin, Ireland, 2017. [Google Scholar]
- Yu, Z.; Morrison, M. Improved Extraction of PCR-Quality Community DNA from Digesta and Fecal Samples. Biotechniques 2004, 36, 808–812. [Google Scholar] [CrossRef]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Hut-tenhower, C.; Segata, N. MetaPhlAn2 for Enhanced Metagenomic Taxonomic Profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Silva, G.G.Z.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: A Tool for Agile Func-tional Analysis of Shotgun Metagenomic Data. Bioinformatics 2015, 32, 354–361. [Google Scholar] [CrossRef]
- Wang, S.; Huang, Y.; Zhou, C.; Wu, H.; Zhao, J.; Wu, L.; Zhao, M.; Zhang, F.; Liu, H. The Role of Autophagy and Related MicroRNAs in Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2018, 2018, 7565076. [Google Scholar] [CrossRef]
- Salaheen, S.; Kim, S.W.; Karns, J.S.; Hovingh, E.; Haley, B.J.; Van Kessel, J.A.S. Metagenomic Analysis of the Fecal Microbiomes from Escherichia Coli O157:H7-Shedding and Non-Shedding Cows on a Single Dairy Farm. Food Control. 2019, 102, 76–80. [Google Scholar] [CrossRef]
- Arrazuria, R.; Elguezabal, N.; Juste, R.A.; Derakhshani, H.; Khafipour, E. Mycobacterium Avium Subspecies Paratuberculosis Infection Modifies Gut Microbiota under Different Dietary Conditions in a Rabbit Model. Front. Microbiol. 2016, 7, 446. [Google Scholar] [CrossRef]
- Turroni, F.; Foroni, E.; Pizzetti, P.; Giubellini, V.; Ribbera, A.; Merusi, P.; Cagnasso, P.; Bizzarri, B.; De’Angelis, G.L.; Shanahan, F.; et al. Exploring the Diversity of the Bifidobacterial Population in the Human Intestinal Tract. Appl. Environ. Microbiol. 2009, 75, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Lamendella, R.; Santo Domingo, J.W.; Kelty, C.; Oerther, D.B. Bifidobacteria in Feces and Envi-ronmental Waters. Appl. Environ. Microbiol. 2008, 74, 575–584. [Google Scholar] [CrossRef]
- Furrie, E.; Macfarlane, S.; Cummings, J.H.; Macfarlane, G.T. Systemic Antibodies towards Mucosal Bacteria in Ulcerative Colitis and Crohn’s Disease Differentially Activate the Innate Immune Response. Gut 2004, 53, 91–98. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- O’Hara, E.; Kelly, A.; McCabe, M.S.; Kenny, D.A.; Guan, L.L.; Waters, S.M. Effect of a Butyr-ate-Fortified Milk Replacer on Gastrointestinal Microbiota and Products of Fermentation in Artifi-cially Reared Dairy Calves at Weaning. Sci. Rep. 2018, 8, 14901. [Google Scholar] [CrossRef]
- Kim, M.; Kuehn, L.A.; Bono, J.L.; Berry, E.D.; Kalchayanand, N.; Freetly, H.C.; Benson, A.K. The Impact of the Bovine Faecal Microbiome on Escherichia Coli O157: H7 Prevalence and Enumeration in Naturally Infected Cattle. J. Appl. Microbiol. 2017, 123, 1027–1042. [Google Scholar] [CrossRef]
- Dill-Mcfarland, K.A.; Breaker, J.D.; Suen, G. Microbial Succession in the Gastrointestinal Tract of Dairy Cows from 2 Weeks to First Lactation. Sci. Rep. 2017, 7, 40864. [Google Scholar] [CrossRef]
- Roussey, J.A.; Steibel, J.P.; Coussens, P.M. Regulatory T Cell Activity and Signs of T Cell Unresponsiveness in Bovine Paratuberculosis. Front. Vet. Sci. 2014, 1, 20. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Insights into the Role of Erysipelotrichaceae in the Human Host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Xu, J.; Leip, D.D.; Chen, C.H.; Westover, B.P.; Weatherford, J.; Buhler, J.D.; Gordon, J.I. Glycan Foraging in Vivo by an Intestine-Adapted Bacterial Symbiont. Science 2005, 307, 1955–1959. [Google Scholar] [CrossRef]
- Curtis, M.M.; Hu, Z.; Klimko, C.; Narayanan, S.; Deberardinis, R.; Sperandio, V. The Gut Com-mensal Bacteroides Thetaiotaomicron Exacerbates Enteric Infection through Modification of the Metabolic Landscape. Cell Host Microbe 2014, 16, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Resta-Lenert, S.; Barrett, K.E. Probiotics and Commensals Reverse TNF-α- and IFN-γ-Induced Dysfunction in Human Intestinal Epithelial Cells. Gastroenterology 2006, 130, 731–746. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | p-Value | Correlation Co-Efficient |
|---|---|---|
| Methanobrevibacter ruminantium | 0.039 | 0.162 |
| Corynebacterium efficiens | 0.007 | 0.211 |
| Bacteroides thetaiotaomicron | 0.045 | 0.157 |
| Enterococcus hirae | 0.007 | 0.209 |
| Butyrivibrio unclassified | 0.005 | 0.220 |
| Erysipelotrichaceae bacterium 21_3 | 0.006 | 0.213 |
| Penicillium chrysogenum | 0.006 | 0.213 |
| Species | p-Value | Correlation Co-Efficient |
|---|---|---|
| Corynebacterium efficiens | 0.048 | −0.319 |
| Bifidobacterium adolescentis | 0.014 | −0.39 |
| Bifidobacterium pseudolongum | 0.025 | 0.357 |
| Alistipes unclassified | 0.033 | 0.342 |
| Desulfovibrio piger | 0.044 | 0.325 |
| Species | p-Values | Correlation Co-Efficient |
|---|---|---|
| Bacteroides thetaiotaomicron | 0.031 | 0.293 |
| Bacteroidales bacterium_ph8 | 0.049 | 0.27 |
| Dorea formicigenerans | 0.012 | 0.34 |
| Faecalibacterium prausnitzii | 0.024 | 0.308 |
| Escherichia unclassified | 0.037 | 0.285 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matthews, C.; Walsh, A.M.; Gordon, S.V.; Markey, B.; Cotter, P.D.; O' Mahony, J. Differences in Faecal Microbiome Taxonomy, Diversity and Functional Potential in a Bovine Cohort Experimentally Challenged with Mycobacterium avium subsp. paratuberculosis (MAP). Animals 2023, 13, 1652. https://doi.org/10.3390/ani13101652
Matthews C, Walsh AM, Gordon SV, Markey B, Cotter PD, O' Mahony J. Differences in Faecal Microbiome Taxonomy, Diversity and Functional Potential in a Bovine Cohort Experimentally Challenged with Mycobacterium avium subsp. paratuberculosis (MAP). Animals. 2023; 13(10):1652. https://doi.org/10.3390/ani13101652
Chicago/Turabian StyleMatthews, Chloe, Aaron M. Walsh, Stephen V. Gordon, Bryan Markey, Paul D. Cotter, and Jim O' Mahony. 2023. "Differences in Faecal Microbiome Taxonomy, Diversity and Functional Potential in a Bovine Cohort Experimentally Challenged with Mycobacterium avium subsp. paratuberculosis (MAP)" Animals 13, no. 10: 1652. https://doi.org/10.3390/ani13101652