Longitudinal Changes in Milk Microorganisms in the First Two Months of Lactation of Primiparous and Multiparous Cows

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Sample Collection
2.3. DNA Extraction and Amplification of V4 Regions of 16S rRNA Gene
2.4. Bioinformatical and Statistical Analysis
2.5. Milk Components Analysis
3. Results
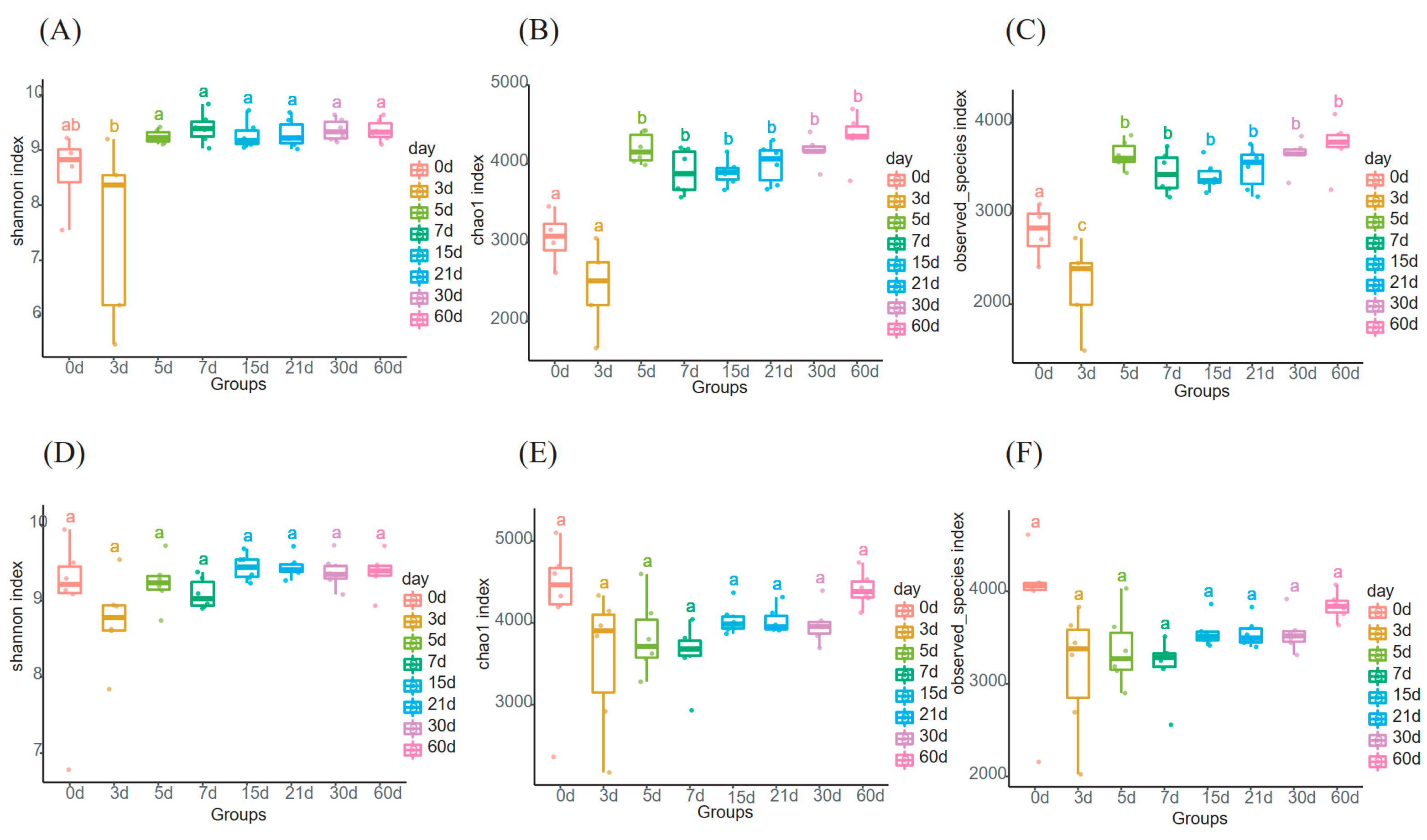
3.1. Alpha Diversity of the Milk Microbiome Community in Primiparous and Multiparous Cows
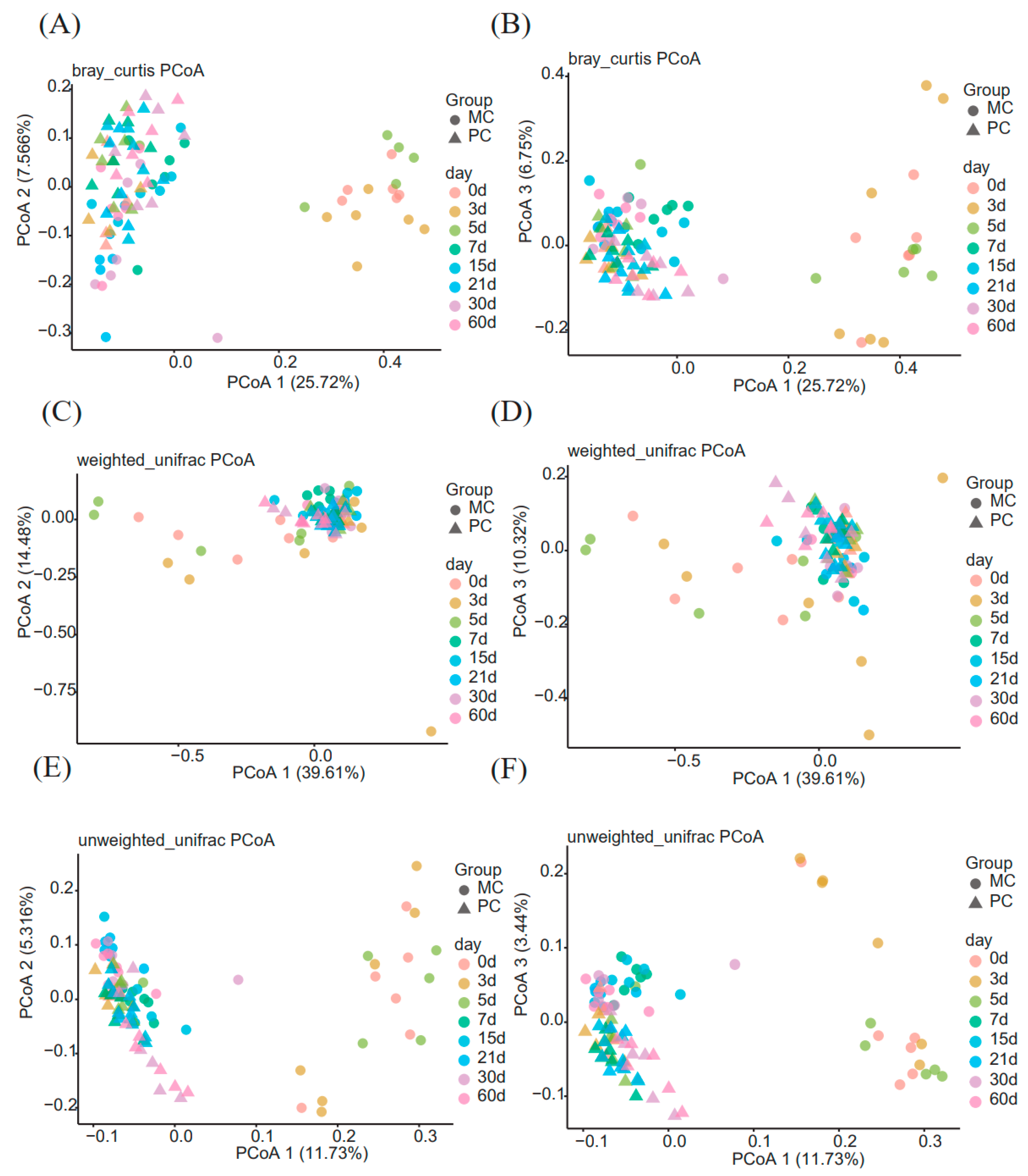
3.2. Beta Diversity of the Milk Microbiome Community in Primiparous and Multiparous Cows
3.3. Composition Changes in the Milk Microbiome Community with DIM in Primiparous and Multiparous Cows
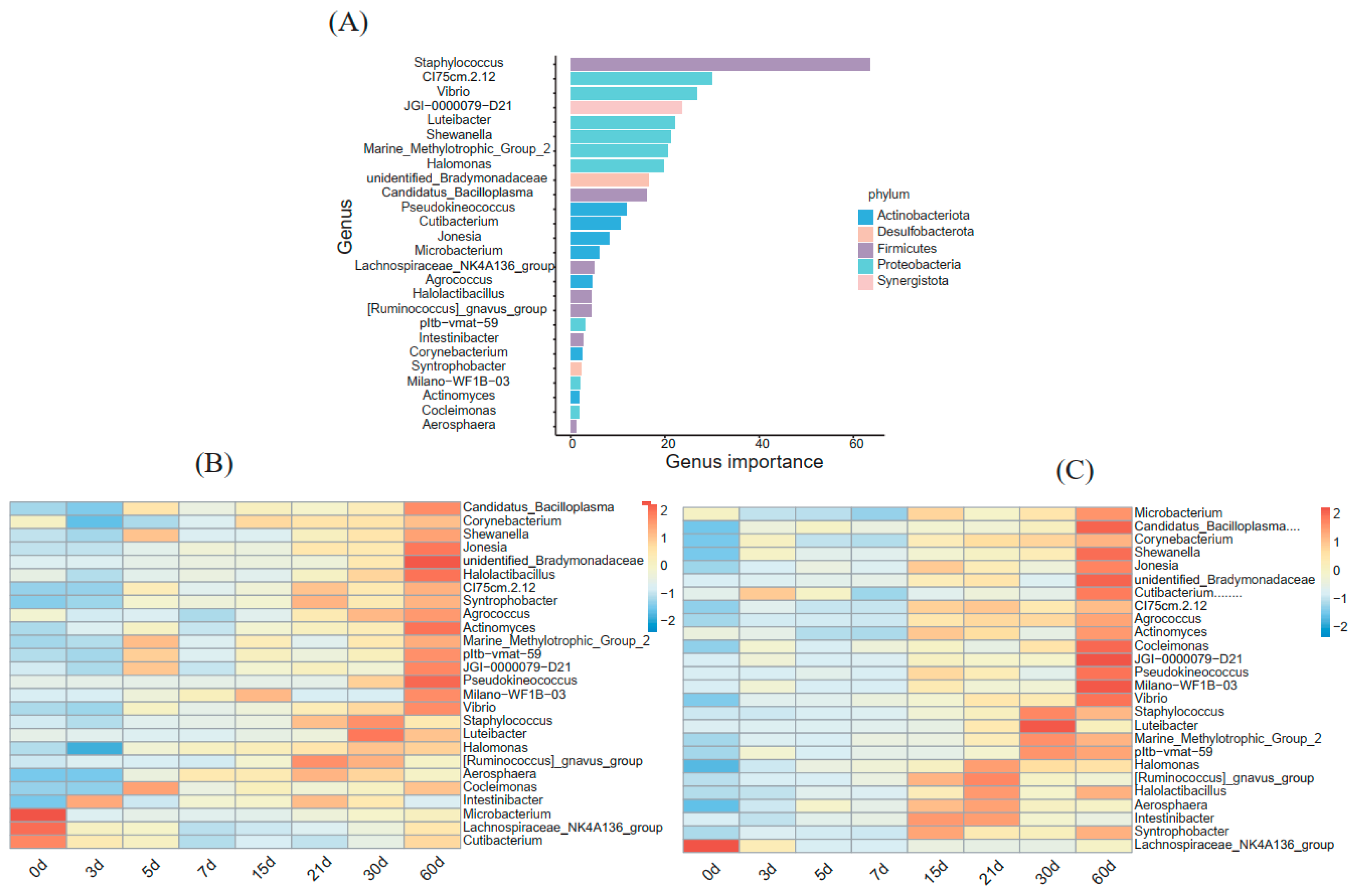
3.4. A Random Forest Model Correlating Milk Bacterial Taxonomic Biomarkers with DIM in Primiparous and Multiparous Cows
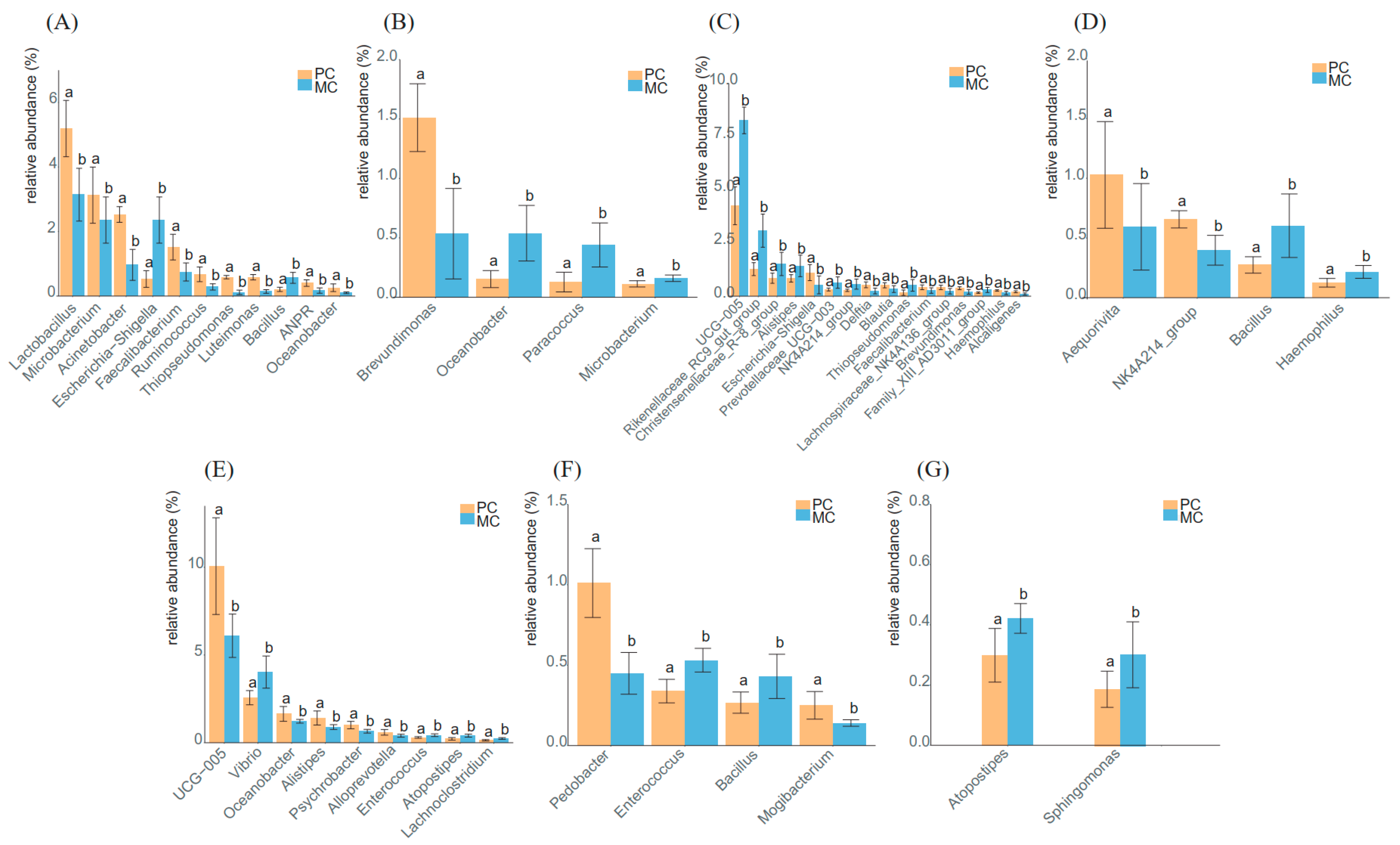
3.5. Analysis of the Predominant Genera in Primiparous and Multiparous Cows
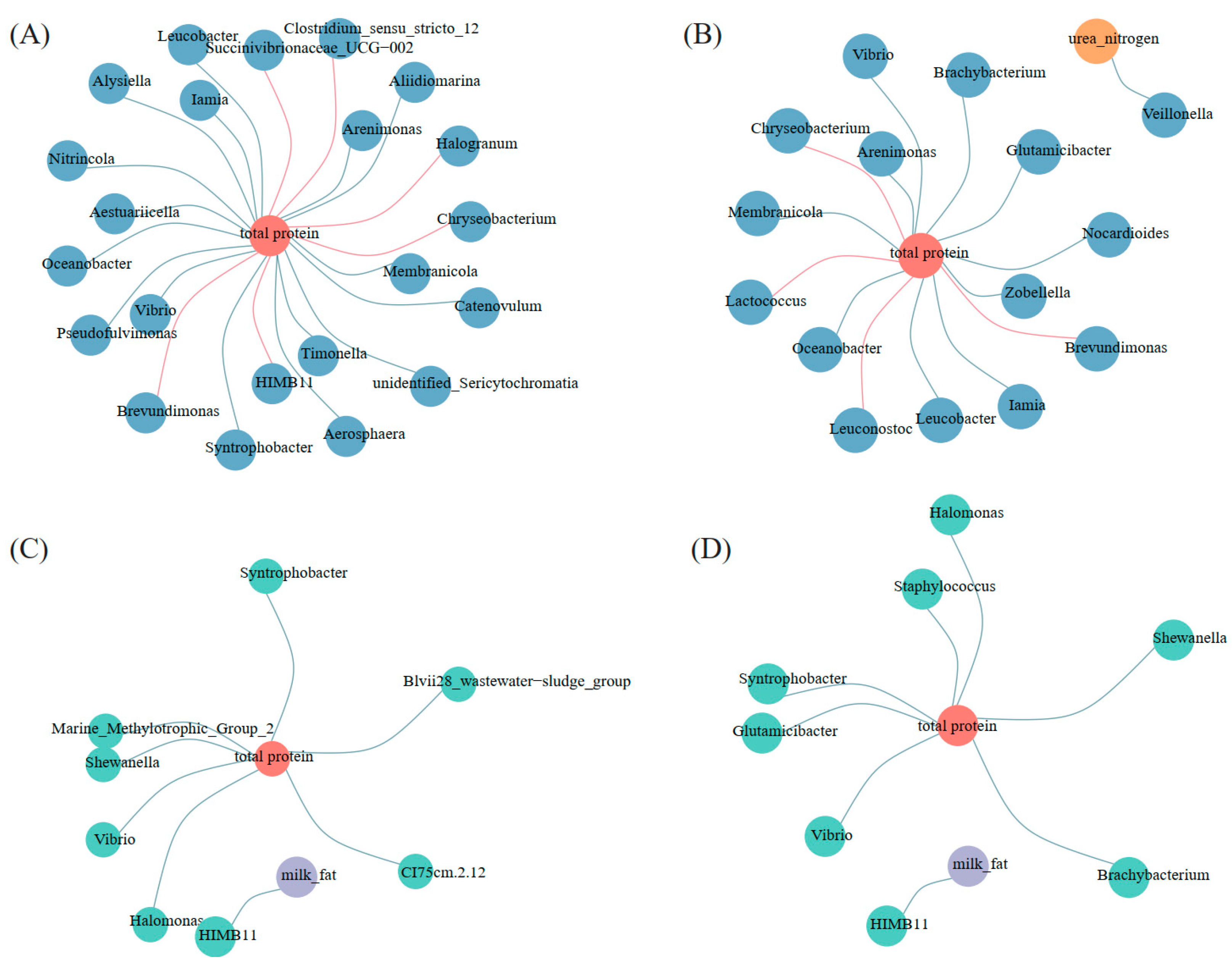
3.6. Co-Occurrence Networks of Characteristic Bacteria and the Components of Milk
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Addis, M.F.; Tanca, A.; Uzzau, S.; Oikonomou, G.; Bicalho, R.C.; Moroni, P. The bovine milk microbiota: Insights and perspectives from -omics studies. Mol. Biosyst. 2016, 12, 2359–2372. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.; Langa, S.; Martín, V.; Maldonado, A.; Jiménez, E.; Martín, R.; Rodríguez, J.M. The human milk microbiota: Origin and potential roles in health and disease. Pharmacol. Res. 2013, 69, 1–10. [Google Scholar] [CrossRef]
- Jeurink, P.V.; Bergenhenegouwen, J.; Jiménez, E.; Knippels, L.M.; Fernández, L.; Garssen, J.; Knol, J.; Rodríguez, J.M.; Martín, R. Human milk: A source of more life than we imagine. Benef. Microbes 2013, 4, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.-W.; Shen, Y.-Y.; Li, X.; Zhang, H.; Niu, H.; Fang, L.-Y.; Xiong, B.-H.; Tong, J.-J.; Jiang, L.-S. Microbiome and metabolic changes of milk in response to dietary supplementation with bamboo leaf extract in dairy cows. Front. Nutr. 2021, 8, 723446. [Google Scholar]
- Zhan, J.-W.; Tong, J.-J.; Xiong, B.-H.; Jiang, L.-S. Research progress on changes of microbial community structure and its influence factors in milk of dairy cows. Chin. J. Anim. Nutr. 2021, 33, 3686–3692. [Google Scholar]
- Stelwagen, K.; Carpenter, E.; Haigh, B.; Hodgkinson, A.; Wheeler, T.T. Immune components of bovine colostrum and milk. J. Anim. Sci. 2009, 87, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, G.; Machado, V.S.; Santisteban, C.; Schukken, Y.H.; Bicalho, R.C. Microbial diversity of bovine mastitic milk as described by pyrosequencing of metagenomic 16s rDNA. PLoS ONE 2012, 7, e47671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomou, G.; Bicalho, M.L.; Meira, E.; Rossi, R.E.; Foditsch, C.; Machado, V.S.; Teixeira, A.G.; Santisteban, C.; Schukken, Y.H.; Bicalho, R.C. Microbiota of cow’s milk; distinguishing healthy, sub-clinically and clinically diseased quarters. PLoS ONE 2014, 9, e85904. [Google Scholar] [CrossRef] [Green Version]
- Malmuthuge, N.; Chen, Y.; Liang, G.; Goonewardene, L.A.; Guan, L. Heat-treated colostrum feeding promotes beneficial bacteria colonization in the small intestine of neonatal calves. J. Dairy Sci. 2015, 98, 8044–8053. [Google Scholar] [CrossRef] [Green Version]
- Lima, S.F.; Teixeira, A.G.V.; Lima, F.S.; Ganda, E.K.; Higgins, C.H.; Oikonomou, G.; Bicalho, R.C. The bovine colostrum microbiome and its association with clinical mastitis. J. Dairy Sci. 2017, 100, 3031–3042. [Google Scholar] [CrossRef] [Green Version]
- Ballas, P.; Pothmann, H.; Pothmann, I.; Drillich, M.; Ehling-Schulz, M.; Wagener, K. Dynamics and diversity of intrauterine anaerobic microbiota in dairy cows with clinical and subclinical endometritis. Animals 2022, 13, 82. [Google Scholar] [CrossRef]
- Quadros, D.L.; Zanella, R.; Bondan, C.; Zanella, G.C.; Facioli, F.L.; da Silva, A.N.; Zanella, E.L. Study of vaginal microbiota of Holstein cows submitted to an estrus synchronization protocol with the use of intravaginal progesterone device. Res. Vet. Sci. 2020, 131, 1–6. [Google Scholar] [CrossRef]
- Pitta, D.W.; Kumar, S.; Vecchiarelli, B.; Shirley, D.J.; Bittinger, K.; Baker, L.D.; Ferguson, J.D.; Thomsen, N. Temporal dynamics in the ruminal microbiome of dairy cows during the transition period. J. Anim. Sci. 2014, 92, 4014–4022. [Google Scholar] [CrossRef] [PubMed]
- Bogado Pascottini, O.; Spricigo, J.F.W.; Van Schyndel, S.J.; Mion, B.; Rousseau, J.; Weese, J.S.; LeBlanc, S.J. Effects of parity, blood progesterone, and non-steroidal anti-inflammatory treatment on the dynamics of the uterine microbiota of healthy postpartum dairy cows. PLoS ONE 2021, 16, e0233943. [Google Scholar] [CrossRef]
- Niyazbekova, Z.; Yao, X.T.; Liu, M.J.; Bold, N.; Tong, J.Z.; Chang, J.J.; Wen, Y.; Li, L.; Wang, Y.; Chen, D.K.; et al. Compositional and functional comparisons of the microbiota in the colostrum and mature milk of dairy goats. Animals 2020, 10, 1955. [Google Scholar] [CrossRef] [PubMed]
- Winther, A.R.; Narvhus, J.A.; Smistad, M.; da Silva Duarte, V.; Bombelli, A.; Porcellato, D. Longitudinal dynamics of the bovine udder microbiota. Anim. Microbiome 2022, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, V.D.; Ahir, V.B.; Koringa, P.G.; Jakhesara, S.J.; Rank, D.N.; Nauriyal, D.S.; Kunjadia, A.P.; Joshi, C.G. Milk microbiome signatures of subclinical mastitis-affected cattle analysed by shotgun sequencing. J. Appl. Microbiol. 2012, 112, 639–650. [Google Scholar] [CrossRef]
- Oikonomou, G.; Addis, M.F.; Chassard, C.; Nader-Macias, M.E.F.; Grant, I.; Delbès, C.; Bogni, I.; Loir, Y.L.; Even, S. Milk microbiota: What are we exactly talking about? Front. Microbiol. 2020, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parigi, S.M.; Eldh, M.; Larssen, P.; Gabrielsson, S.; Villablanca, E.J. Breast milk and solid food shaping intestinal immunity. Front. Immunol. 2015, 6, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, M.; Xie, X.; Bao, H.; Sun, L.; He, T.; Zhao, H.; Zhou, Y.; Zhang, L.; Zhang, H.; Wei, R.; et al. Insights into the bovine milk microbiota in dairy farms with different incidence rates of subclinical mastitis. Front. Microbiol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.X.; Lima, S.F.; Canniatti-Brazaca, S.G.; Bicalho, R.C. The microbiome of bulk tank milk: Characterization and associations with somatic cell count and bacterial count. J. Dairy Sci. 2017, 100, 2536–2552. [Google Scholar] [CrossRef] [Green Version]
- Derakhshani, H.; Fehr, K.B.; Sepehri, S.; Francoz, D.; De Buck, J.; Barkema, H.W.; Plaizier, J.C.; Khafipour, E. Invited review: Microbiota of the bovine udder: Contributing factors and potential implications for udder health and mastitis susceptibility. J. Dairy Sci. 2018, 101, 10605–10625. [Google Scholar] [CrossRef] [Green Version]
- Kable, M.E.; Srisengfa, Y.; Laird, M.; Zaragoza, J.; McLeod, J.; Heidenreich, J.; Marco, M.L. The core and seasonal microbiota of raw bovine milk in tanker trucks and the impact of transfer to a milk processing facility. MBio 2016, 7, e00836-16. [Google Scholar] [CrossRef] [Green Version]
- Maity, S.; Ambatipudi, K. Mammary microbial dysbiosis leads to the zoonosis of bovine mastitis: A One-Health perspective. FEMS Microbiol. Ecol. 2020, 97, fiaa241. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.R.; Callaway, T.R.; Lourenco, J.M.; Ryman, V.E. Characterization of rumen, fecal, and milk microbiota in lactating dairy cows. Front. Microbiol. 2022, 13, 984119. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Cossart, P. Prokaryotic transcriptomics: A new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 2010, 11, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Curley, D.; O’Callaghan, T.F.; O’Shea, C.A.; Dempsey, E.M.; O’Toole, P.W.; Ross, R.P.; Ryan, C.A.; Stanton, C. The composition of human milk and infant faecal microbiota over the first three months of life: A pilot study. Sci. Rep. 2017, 7, 40597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, T.L.; Hosid, S.; Ioshikhes, I.; Altosaar, I. Human milk metagenome: A functional capacity analysis. BMC Microbiol. 2013, 13, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.H.; Vaidya, Y.H.; Patel, R.J.; Pandit, R.J.; Joshi, C.G.; Kunjadiya, A.P. Culture independent assessment of human milk microbial community in lactational mastitis. Sci. Rep. 2017, 7, 7804. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Huo, W.; Zhu, W.; Mao, S. Characterization of bacterial community of raw milk from dairy cows during subacute ruminal acidosis challenge by high-throughput sequencing. J. Sci. Food Agric. 2015, 95, 1072–1079. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Morgan, X.C.; Segata, N.; Waldron, L.; Reyes, J.; Earl, A.M.; Giannoukos, G.; Boylan, M.R.; Ciulla, D.; Gevers, D.; et al. Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 2014, 111, E2329–E2338. [Google Scholar] [CrossRef] [Green Version]
- Gurung, M.; Nam, H.M.; Tamang, M.D.; Chae, M.H.; Jang, G.C.; Jung, S.C.; Lim, S.K. Prevalence and antimicrobial susceptibility of Acinetobacter from raw bulk tank milk in Korea. J. Dairy Sci. 2013, 96, 1997–2002. [Google Scholar] [CrossRef]
- Ge, Y.; Zhu, W.; Chen, L.; Li, D.; Li, Q.; Jie, H. The maternal milk microbiome in mammals of different types and its potential role in the neonatal gut microbiota composition. Animals 2021, 11, 3349. [Google Scholar] [CrossRef]
- Mtshali, K.; Khumalo, Z.T.H.; Kwenda, S.; Arshad, I.; Thekisoe, O.M.M. Exploration and comparison of bacterial communities present in bovine faeces, milk and blood using 16S rRNA metagenomic sequencing. PLoS ONE 2022, 17, e0273799. [Google Scholar] [CrossRef] [PubMed]
- Cendron, F.; Niero, G.; Carlino, G.; Penasa, M.; Cassandro, M. Characterizing the fecal bacteria and archaea community of heifers and lactating cows through 16S rRNA next-generation sequencing. J. Appl. Genet. 2020, 61, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liao, M.; Zhou, N.; Bao, L.; Ma, K.; Zheng, Z.; Wang, Y.; Liu, C.; Wang, W.; Wang, J.; et al. Parabacteroides distasonis alleviates obesity and metabolic dysfunctions via production of succinate and secondary bile acids. Cell Rep. 2019, 26, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Hou, K.; Tong, J.; Zhang, H.; Gao, S.; Guo, Y.; Niu, H.; Xiong, B.; Jiang, L. Microbiome and metabolic changes in milk in response to artemisinin supplementation in dairy cows. AMB Express 2020, 10, 154. [Google Scholar] [CrossRef]
- Young, W.; Hine, B.C.; Wallace, O.A.; Callaghan, M.; Bibiloni, R. Transfer of intestinal bacterial components to mammary secretions in the cow. PeerJ 2015, 3, e888. [Google Scholar] [CrossRef] [PubMed]
- Scarsella, E.; Zecconi, A.; Cintio, M.; Stefanon, B. Characterization of microbiome on feces, blood and milk in dairy cows with different milk leucocyte pattern. Animals 2021, 11, 1463. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Wang, Y.; Kang, X.; Liu, P.; Wang, G. Research progress on the association between mastitis and gastrointestinal microbes in dairy cows and the effect of probiotics. Microb. Pathog. 2022, 173, 105809. [Google Scholar] [CrossRef]
- Schwarz, D.G.G.; Shoyama, F.M.; Oliveira, L.L.; Sreevatsan, S.; Moreira, M.A.S. Rapid baso-apical translocation of Mycobacterium avium ssp. paratuberculosis in mammary epithelial cells in the presence of Escherichia coli. J. Dairy Sci. 2018, 101, 6287–6295. [Google Scholar] [CrossRef] [Green Version]
- Gärtner, T.; Gernand, E.; Gottschalk, J.; Donat, K. Relationships between body condition, body condition loss, and serum metabolites during the transition period in primiparous and multiparous cows. J. Dairy Sci. 2019, 102, 9187–9199. [Google Scholar] [CrossRef]
- Hu, X.; Li, S.; Fu, Y.; Zhang, N. Targeting gut microbiota as a possible therapy for mastitis. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1409–1423. [Google Scholar] [CrossRef]
- Mbuthia, O.W. Diarrheal correlates associated with enteric bacterial infections among children below five years in Murang’a County, Kenya. Pan. Afr. Med. J. 2019, 34, 170. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.T.; Lacroix, C.; Braegger, C.P.; Chassard, C. Early colonization of functional groups of microbes in the infant gut. Environ. Microbiol. 2016, 18, 2246–2258. [Google Scholar] [CrossRef]
- Pham, V.T.; Lacroix, C.; Braegger, C.P.; Chassard, C. Lactate-utilizing community is associated with gut microbiota dysbiosis in colicky infants. Sci. Rep. 2017, 7, 11176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koltes, D.A.; Spurlock, D.M. Adipose tissue angiopoietin-like protein 4 messenger RNA changes with altered energy balance in lactating Holstein cows. Domest. Anim. Endocrinol. 2012, 43, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Pinzón-Sánchez, C.; Ruegg, P.L. Risk factors associated with short-term post-treatment outcomes of clinical mastitis. J. Dairy Sci. 2011, 94, 3397–3410. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Zhang, C.; Wang, C.; Tan, Z. Intestinal mucosal bacterial diversity of antibiotic-associated diarrhea (AAD) mice treated with Debaryomyces hansenii and Qiweibaizhu powder. 3 Biotech 2020, 10, 392. [Google Scholar] [CrossRef] [PubMed]
- Rackaityte, E.; Halkias, J.; Fukui, E.M.; Mendoza, V.F.; Hayzelden, C.; Crawford, E.D.; Fujimura, K.E.; Burt, T.D.; Lynch, S.V. Viable bacterial colonization is highly limited in the human intestine in utero. Nat. Med. 2020, 26, 599–607. [Google Scholar] [CrossRef]
- Quercia, S.; Freccero, F.; Castagnetti, C.; Soverini, M.; Turroni, S.; Biagi, E.; Rampelli, S.; Lanci, A.; Mariella, J.; Chinellato, E.; et al. Early colonisation and temporal dynamics of the gut microbial ecosystem in Standardbred foals. Equine Vet. J. 2019, 51, 231–237. [Google Scholar] [CrossRef]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef] [Green Version]
- Derakhshani, H.; Plaizier, J.C.; De Buck, J.; Barkema, H.W.; Khafipour, E. Association of bovine major histocompatibility complex (BoLA) gene polymorphism with colostrum and milk microbiota of dairy cows during the first week of lactation. Microbiome 2018, 6, 203. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, E.; de Andrés, J.; Manrique, M.; Pareja-Tobes, P.; Tobes, R.; Martínez-Blanch, J.F.; Codoñer, F.M.; Ramón, D.; Fernández, L.; Rodríguez, J.M. Metagenomic analysis of milk of healthy and mastitis-suffering women. J. Hum. Lact. 2015, 31, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Boix-Amorós, A.; Collado, M.C.; Mira, A. Relationship between milk microbiota, bacterial load, macronutrients, and human cells during lactation. Front. Microbiol. 2016, 7, 492. [Google Scholar] [CrossRef] [Green Version]
- Urbaniak, C.; Angelini, M.; Gloor, G.B.; Reid, G. Human milk microbiota profiles in relation to birthing method, gestation and infant gender. Microbiome 2016, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, L.; Arroyo, R.; Espinosa, I.; Marín, M.; Jiménez, E.; Rodríguez, J.M. Probiotics for human lactational mastitis. Benef. Microbes 2014, 5, 169–183. [Google Scholar] [CrossRef]
- Carson, D.A.; Barkema, H.W.; Naushad, S.; De Buck, J. Bacteriocins of non-aureus Staphylococci isolated from bovine milk. Appl. Environ. Microbiol. 2017, 83, e01015–e01017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jost, T.; Lacroix, C.; Braegger, C.P.; Rochat, F.; Chassard, C. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ. Microbiol. 2014, 16, 2891–2904. [Google Scholar] [CrossRef]
- Cerdó, T.; Ruiz, A.; Acuña, I.; Jáuregui, R.; Jehmlich, N.; Haange, S.B.; von Bergen, M.; Suárez, A.; Campoy, C. Gut microbial functional maturation and succession during human early life. Environ. Microbiol. 2018, 20, 2160–2177. [Google Scholar] [CrossRef] [PubMed]
- Nogacka, A.M.; Salazar, N.; Arboleya, S.; Suárez, M.; Fernández, N.; Solís, G.; de Los Reyes-Gavilán, C.G.; Gueimonde, M. Early microbiota, antibiotics and health. Cell. Mol. Life. Sci. 2018, 75, 83–91. [Google Scholar] [CrossRef]
- Röttjers, L.; Faust, K. From hairballs to hypotheses-biological insights from microbial networks. FEMS Microbiol. Rev. 2018, 42, 761–780. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Value |
|---|---|
| Ingredient | % |
| Corn silage | 39.52 |
| Ground corn | 21.83 |
| Alfalfa hay | 7.45 |
| Cottonseed | 3.81 |
| DDGS (distiller dried grains with solubles) | 3.41 |
| Brewers grain | 10.77 |
| Soybean meal | 5.34 |
| Cottonseed protein | 1.37 |
| Soybean hulls | 2.98 |
| Cottonseed meal | 0.85 |
| Premix2 | 1.17 |
| Limestone | 0.7 |
| Choline | 0.05 |
| Soda | 0.75 |
| Total | 100 |
| Nutrient levels | |
| Dry Matter in Dairy Ration (DM, %) | 57.47 |
| NEL3, MJ/kg | 6.72 |
| Crude Protein (CP, %) | 14.80 |
| Crude Fat (EE, %) | 3.77 |
| Crude Fiber (CF, %) | 15.27 |
| Neutral Detergent Fiber (NDF, %) | 34.46 |
| Acid detergent fiber (ADF, %) | 21.34 |
| Ash, % | 4.85 |
| Ca, % | 0.52 |
| P, % | 0.37 |
| Group | Phylum | 0 d | 3 d | 5 d | 7 d | 15 d | 21 d | 30 d | 60 d |
|---|---|---|---|---|---|---|---|---|---|
| PC | Proteobacteria | 28.93 | 34.32 | 39.16 | 31.14 | 37.41 | 33.09 | 33.65 | 34.30 |
| Firmicutes | 28.88 | 22.55 | 26.82 | 36.86 | 30.95 | 29.27 | 30.23 | 30.30 | |
| Actinobacteriota | 5.30 | 3.58 | 8.88 | 7.62 | 11.06 | 15.19 | 13.73 | 12.59 | |
| Bacteroidota | 16.79 | 27.86 | 13.83 | 14.61 | 11.45 | 12.91 | 12.10 | 11.85 | |
| unidentified_Bacteria | 2.18 | 1.50 | 2.19 | 2.58 | 2.08 | 2.22 | 2.20 | 2.50 | |
| Cyanobacteria | 0.97 | 1.86 | 0.64 | 0.44 | 0.52 | 0.77 | 1.19 | 0.87 | |
| Desulfobacterota | 0.34 | 0.22 | 1.31 | 0.65 | 0.48 | 0.45 | 0.50 | 0.66 | |
| Verrucomicrobiota | 0.39 | 0.34 | 0.60 | 0.49 | 0.48 | 0.35 | 0.36 | 0.34 | |
| MC | Proteobacteria | 29.19 | 36.87 | 33.71 | 39.13 | 31.56 | 38.79 | 33.58 | 36.71 |
| Firmicutes | 31.60 | 24.08 | 30.11 | 29.44 | 35.30 | 28.45 | 33.57 | 30.31 | |
| Bacteroidota | 10.42 | 19.26 | 18.61 | 15.40 | 11.45 | 11.99 | 12.67 | 11.78 | |
| Actinobacteriota | 2.75 | 7.30 | 7.72 | 6.95 | 13.03 | 10.65 | 11.46 | 9.79 | |
| unidentified_Bacteria | 1.90 | 1.99 | 2.60 | 1.92 | 2.49 | 2.24 | 2.18 | 2.50 | |
| Cyanobacteria | 14.47 | 0.82 | 0.51 | 0.44 | 0.31 | 0.76 | 0.46 | 0.87 | |
| Desulfobacterota | 0.49 | 0.96 | 0.74 | 0.90 | 0.51 | 0.57 | 0.47 | 0.72 | |
| Verrucomicrobiota | 0.51 | 0.64 | 0.69 | 0.74 | 0.29 | 0.32 | 0.30 | 0.34 | |
| Deinococcota | 0.07 | 0.24 | 0.31 | 0.29 | 0.31 | 0.52 | 0.26 | 0.28 |
| Group | Milk Component | 0 d | 3 d | 5 d | 7 d | 15 d | 21 d | 30 d | 60 d |
|---|---|---|---|---|---|---|---|---|---|
| PC | Milk Fat (%) | 4.41 ± 0.72 | 2.38 ± 1.21 | 5.11 ± 0.861 | 5.19 ± 0.79 | 4.45 ± 0.83 | 4.58 ± 0.52 | 4.06 ± 1.15 | 3.72 ± 0.09 |
| Total Protein (%) | 14.59 ± 1.14 | 6.29 ± 2.54 | 3.40 ± 0.26 | 3.27 ± 0.17 | 3.29 ± 0.22 | 3.03 ± 0.16 | 3.04 ± 0.28 | 3.05 ± 0.25 | |
| Urea Nitrogen (mg/dL) | 16.98 ± 3.12 | 13.95 ± 3.75 | 12.20 ± 3.35 | 12.63 ± 1.97 | 11.18 ± 3.22 | 11.16 ± 2.58 | 11.93 ± 2.82 | 12.17 ± 1.28 | |
| MC | Milk Fat (%) | 4.16 ± 0.49 | 3.24 ± 1.52 | 5.39 ± 0.63 | 5.27 ± 0.39 | 4.82 ± 0.36 | 4.97 ± 0.25 | 3.84 ± 0.07 | 3.99 ± 0.50 |
| Total Protein (%) | 16.56 ± 1.62 | 4.29 ± 1.32 | 4.00 ± 0.42 | 3.73 ± 0.18 | 3.14 ± 0.17 | 3.14 ± 0.19 | 3.10 ± 0.11 | 3.14 ± 0.28 | |
| Urea Nitrogen (mg/dl) | 15.11 ± 4.11 | 13.13 ± 2.22 | 12.36 ± 2.49 | 12.31 ± 2.99 | 12.29 ± 2.66 | 12.71 ± 2.89 | 13.07 ± 2.36 | 14.20 ± 2.82 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Miao, R.; Tao, X.; Wu, J.; Liu, L.; Qu, J.; Liu, H.; Sun, Y.; Li, L.; Qu, Y. Longitudinal Changes in Milk Microorganisms in the First Two Months of Lactation of Primiparous and Multiparous Cows. Animals 2023, 13, 1923. https://doi.org/10.3390/ani13121923
Zhu H, Miao R, Tao X, Wu J, Liu L, Qu J, Liu H, Sun Y, Li L, Qu Y. Longitudinal Changes in Milk Microorganisms in the First Two Months of Lactation of Primiparous and Multiparous Cows. Animals. 2023; 13(12):1923. https://doi.org/10.3390/ani13121923
Chicago/Turabian StyleZhu, Huan, Renfang Miao, Xinxu Tao, Jianhao Wu, Licheng Liu, Jiachen Qu, Hongzhi Liu, Yanting Sun, Lingyan Li, and Yongli Qu. 2023. "Longitudinal Changes in Milk Microorganisms in the First Two Months of Lactation of Primiparous and Multiparous Cows" Animals 13, no. 12: 1923. https://doi.org/10.3390/ani13121923
APA StyleZhu, H., Miao, R., Tao, X., Wu, J., Liu, L., Qu, J., Liu, H., Sun, Y., Li, L., & Qu, Y. (2023). Longitudinal Changes in Milk Microorganisms in the First Two Months of Lactation of Primiparous and Multiparous Cows. Animals, 13(12), 1923. https://doi.org/10.3390/ani13121923