
Application of Nanopore Sequencing for High Throughput Genotyping in Horses
 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Material and DOP-PCR
2.2. Nanopore Library Construction and Sequencing
2.3. Data Analysis
2.4. Population Genetics
2.5. SNPs Validation
3. Results
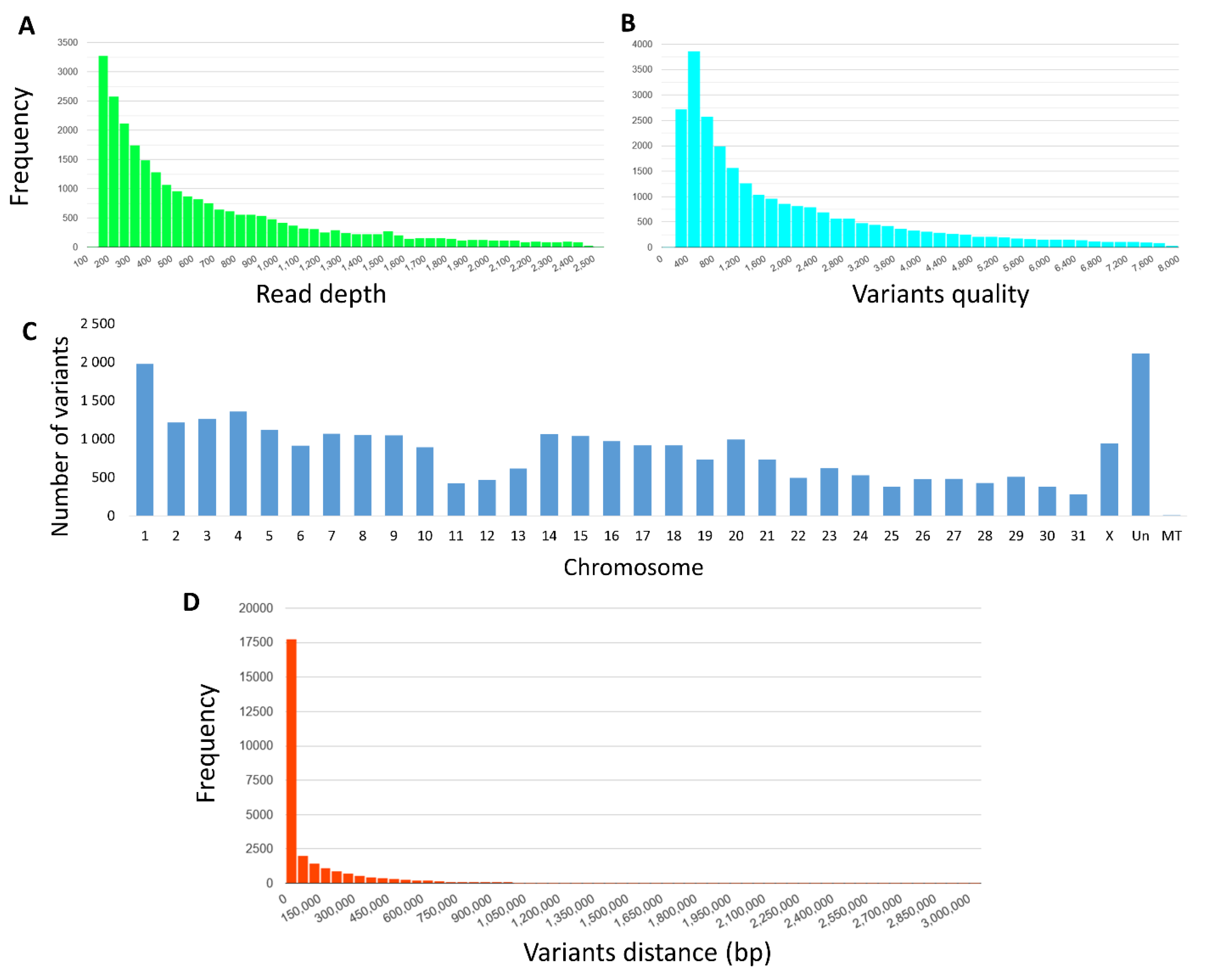
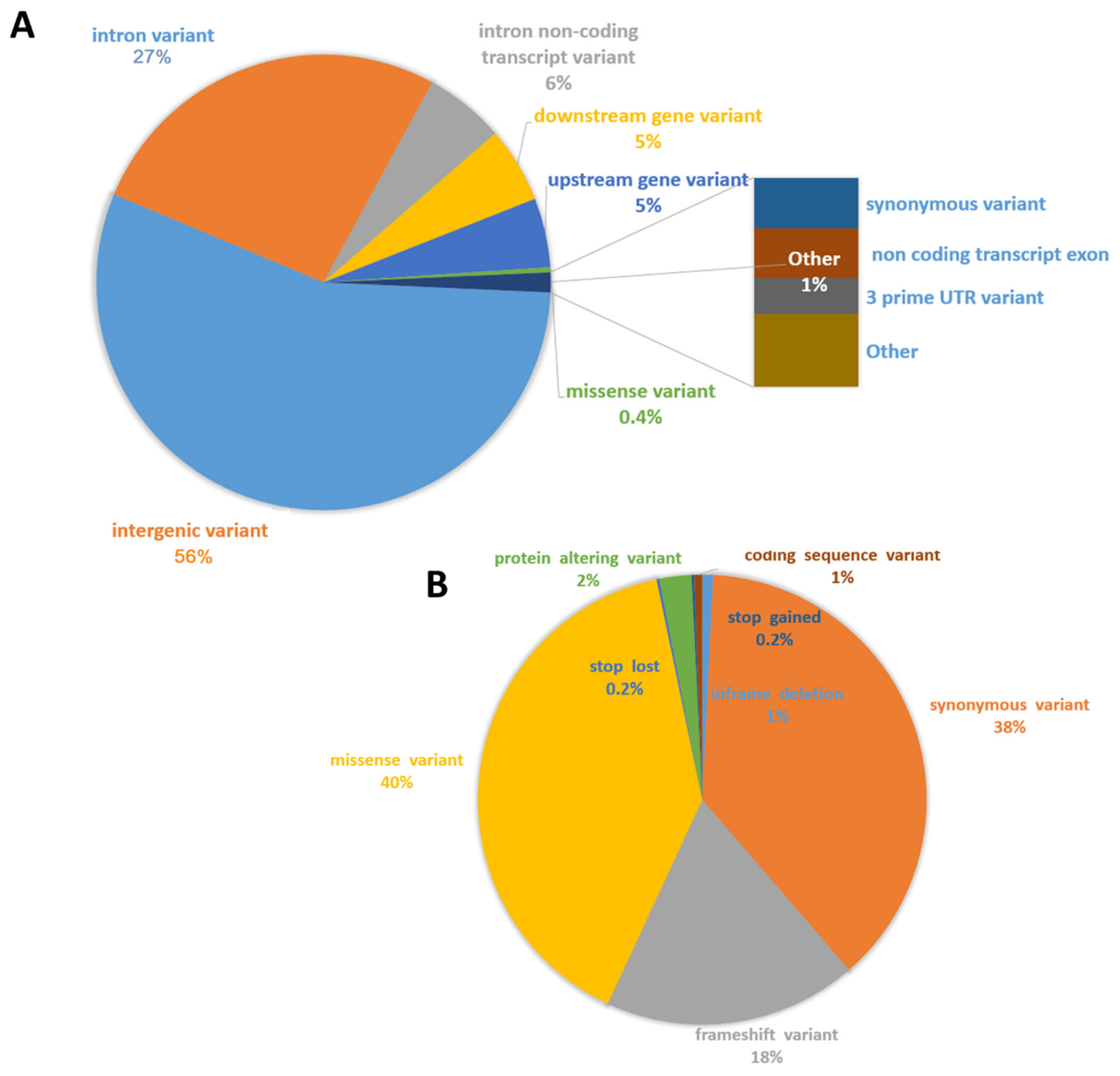
3.1. Sequencing and Variants Discovery
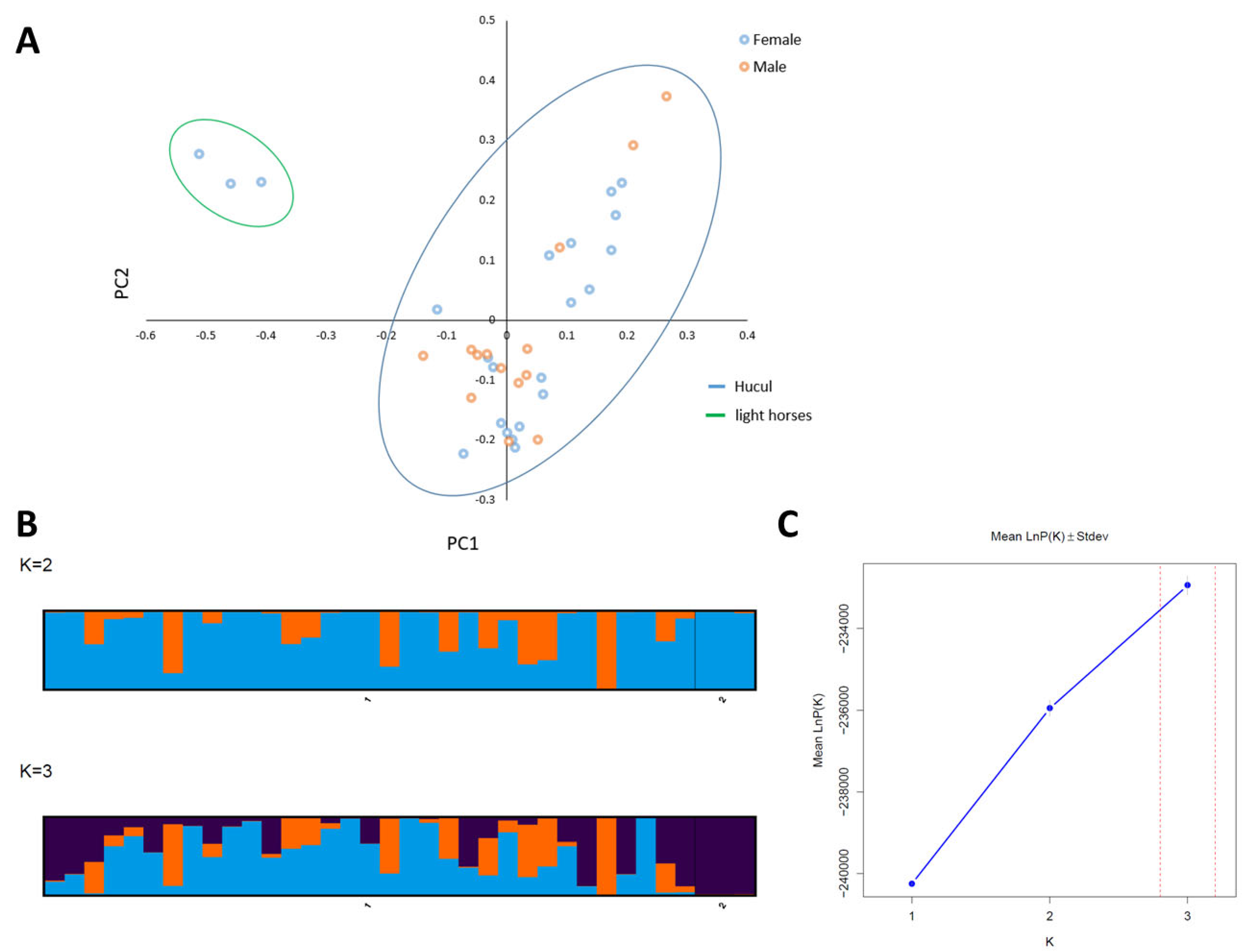
3.2. Application of Variants for Population Genetics
3.3. Variants Detection and Genotyping Accuracy—Validation Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef] [PubMed]
- Ari, Ş.; Arikan, M. Next-Generation Sequencing: Advantages, Disadvantages, and Future BT—Plant Omics: Trends and Applications; Hakeem, K.R., Tombuloğlu, H., Tombuloğlu, G., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 109–135. ISBN 978-3-319-31703-8. [Google Scholar]
- Athanasopoulou, K.; Boti, M.A.; Adamopoulos, P.G.; Skourou, P.C.; Scorilas, A. Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life 2022, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- De Donato, M.; Peters, S.O.; Mitchell, S.E.; Hussain, T.; Imumorin, I.G. Genotyping-by-Sequencing (GBS): A Novel, Efficient and Cost-Effective Genotyping Method for Cattle Using Next-Generation Sequencing. PLoS ONE 2013, 8, e62137. [Google Scholar] [CrossRef]
- Gurgul, A.; Miksza-Cybulska, A.; Szmatoła, T.; Jasielczuk, I.; Piestrzyńska-Kajtoch, A.; Fornal, A.; Semik-Gurgul, E.; Bugno-Poniewierska, M. Genotyping-by-sequencing performance in selected livestock species. Genomics 2018, 111, 186–195. [Google Scholar] [CrossRef]
- Telenius, H.; Carter, N.P.; Bebb, C.E.; Nordenskjöld, M.; Ponder, B.A.J.; Tunnacliffe, A. Degenerate oligonucleotide-primed PCR: General amplification of target DNA by a single degenerate primer. Genomics 1992, 13, 718–725. [Google Scholar] [CrossRef]
- Goddard, M.E.; Hayes, B.J. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 2009, 10, 381–391. [Google Scholar] [CrossRef]
- Zhang, H.; Sachdev, P.S.; Wen, W.; Kochan, N.A.; Crawford, J.D.; Brodaty, H.; Slavin, M.J.; Reppermund, S.; Draper, B.; Zhu, W.; et al. Gray matter atrophy patterns of mild cognitive impairment subtypes. J. Neurol. Sci. 2012, 315, 26–32. [Google Scholar] [CrossRef]
- Szmatoła, T.; Gurgul, A.; Ropka-Molik, K.; Jasielczuk, I.; Zabek, T.; Bugno-Poniewierska, M. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livest. Sci. 2016, 188, 72–80. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Panetto, J.C.d.C.; Machado, M.A.; da Silva, M.V.G.B.; Barbosa, R.S.; dos Santos, G.G.; Leite, R.d.M.H.; Peixoto, M.G.C.D. Parentage assignment using SNP markers, inbreeding and population size for the Brazilian Red Sindhi cattle. Livest. Sci. 2017, 204, 33–38. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.P.; Marth, G.T. Haplotype-based variant detection from short-read sequencing. arXiv 2012. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2013, 14, 209–214. [Google Scholar] [CrossRef]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-L.; Liu, J.-X. StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2018, 18, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kõressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef] [Green Version]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Lamb, H.J.; Hayes, B.J.; Randhawa, I.A.S.; Nguyen, L.T.; Ross, E.M. Genomic prediction using low-coverage portable Nanopore sequencing. PLoS ONE 2021, 16, e0261274. [Google Scholar] [CrossRef]
- Delahaye, C.; Nicolas, J. Sequencing DNA with nanopores: Troubles and biases. PLoS ONE 2021, 16, e0257521. [Google Scholar] [CrossRef]
- Sahlin, K.; Medvedev, P. Error correction enables use of Oxford Nanopore technology for reference-free transcriptome analysis. Nat. Commun. 2021, 12, 2. [Google Scholar] [CrossRef]
- Dorfner, M.; Ott, T.; Ott, P.; Oberprieler, C. Long-read genotyping with SLANG (Simple Long-read loci Assembly of Nanopore data for Genotyping). Appl. Plant Sci. 2022, 10, e11484. [Google Scholar] [CrossRef]
- Malmberg, M.M.; Spangenberg, G.C.; Daetwyler, H.D.; Cogan, N.O.I. Assessment of low-coverage nanopore long read sequencing for SNP genotyping in doubled haploid canola (Brassica napus L.). Sci. Rep. 2019, 9, 8688. [Google Scholar] [CrossRef] [Green Version]
- Brouard, J.-S.; Boyle, B.; Ibeagha-Awemu, E.M.; Bissonnette, N. Low-depth genotyping-by-sequencing (GBS) in a bovine population: Strategies to maximize the selection of high quality genotypes and the accuracy of imputation. BMC Genet. 2017, 18, 32. [Google Scholar] [CrossRef] [Green Version]
- Jasielczuk, I.; Gurgul, A.; Szmatoła, T.; Semik-Gurgul, E.; Pawlina-Tyszko, K.; Stefaniuk-Szmukier, M.; Polak, G.; Tomczyk-Wrona, I.; Bugno-Poniewierska, M. Linkage disequilibrium, haplotype blocks and historical effective population size in Arabian horses and selected Polish native horse breeds. Livest. Sci. 2020, 239, 104095. [Google Scholar] [CrossRef]
- Mackowski, M.; Mucha, S.; Cholewinski, G.; Cieslak, J. Genetic diversity in Hucul and Polish primitive horse breeds. Arch. Anim. Breed. 2015, 58, 23–31. [Google Scholar] [CrossRef]
- Tabata, Y.; Matsuo, Y.; Fujii, Y.; Ohta, A.; Hirota, K. Rapid detection of single nucleotide polymorphisms using the MinION nanopore sequencer: A feasibility study for perioperative precision medicine. JA Clin. Rep. 2022, 8, 17. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurgul, A.; Jasielczuk, I.; Szmatoła, T.; Sawicki, S.; Semik-Gurgul, E.; Długosz, B.; Bugno-Poniewierska, M. Application of Nanopore Sequencing for High Throughput Genotyping in Horses. Animals 2023, 13, 2227. https://doi.org/10.3390/ani13132227
Gurgul A, Jasielczuk I, Szmatoła T, Sawicki S, Semik-Gurgul E, Długosz B, Bugno-Poniewierska M. Application of Nanopore Sequencing for High Throughput Genotyping in Horses. Animals. 2023; 13(13):2227. https://doi.org/10.3390/ani13132227
Chicago/Turabian StyleGurgul, Artur, Igor Jasielczuk, Tomasz Szmatoła, Sebastian Sawicki, Ewelina Semik-Gurgul, Bogusława Długosz, and Monika Bugno-Poniewierska. 2023. "Application of Nanopore Sequencing for High Throughput Genotyping in Horses" Animals 13, no. 13: 2227. https://doi.org/10.3390/ani13132227
APA StyleGurgul, A., Jasielczuk, I., Szmatoła, T., Sawicki, S., Semik-Gurgul, E., Długosz, B., & Bugno-Poniewierska, M. (2023). Application of Nanopore Sequencing for High Throughput Genotyping in Horses. Animals, 13(13), 2227. https://doi.org/10.3390/ani13132227