

Establishing a Sequencing Method for the Whole Mitochondrial DNA of Domestic Dogs

 , , ,
, , ,
Abstract
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Research Ethics
2.2. Dogs and Collected Specimens
2.3. Extraction of Oral Mucosa DNA
2.4. Design of Primer Pairs for mtDNA
2.5. Conditions of the LR-PCR
2.6. Library Preparation of NGS
2.7. NGS Rum
2.8. Bioinformatics Analysis
3. Results
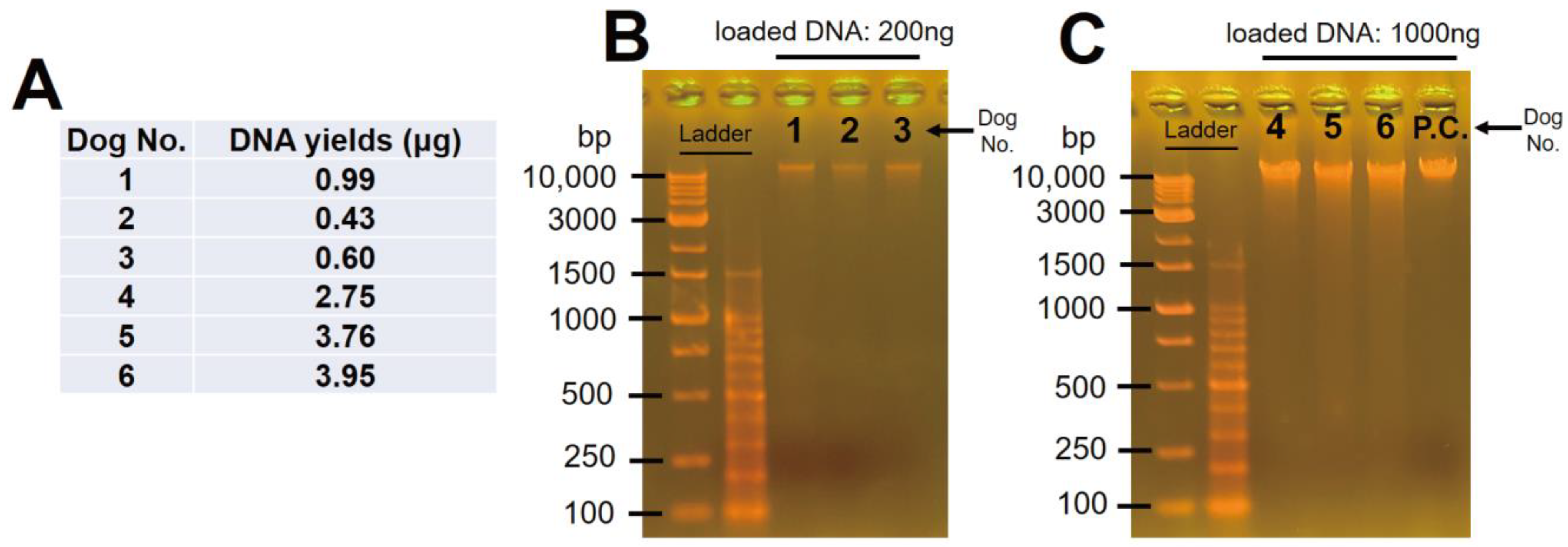
3.1. Oral Mucosal DNA Was Successfully Extracted from and Purified in All Samples
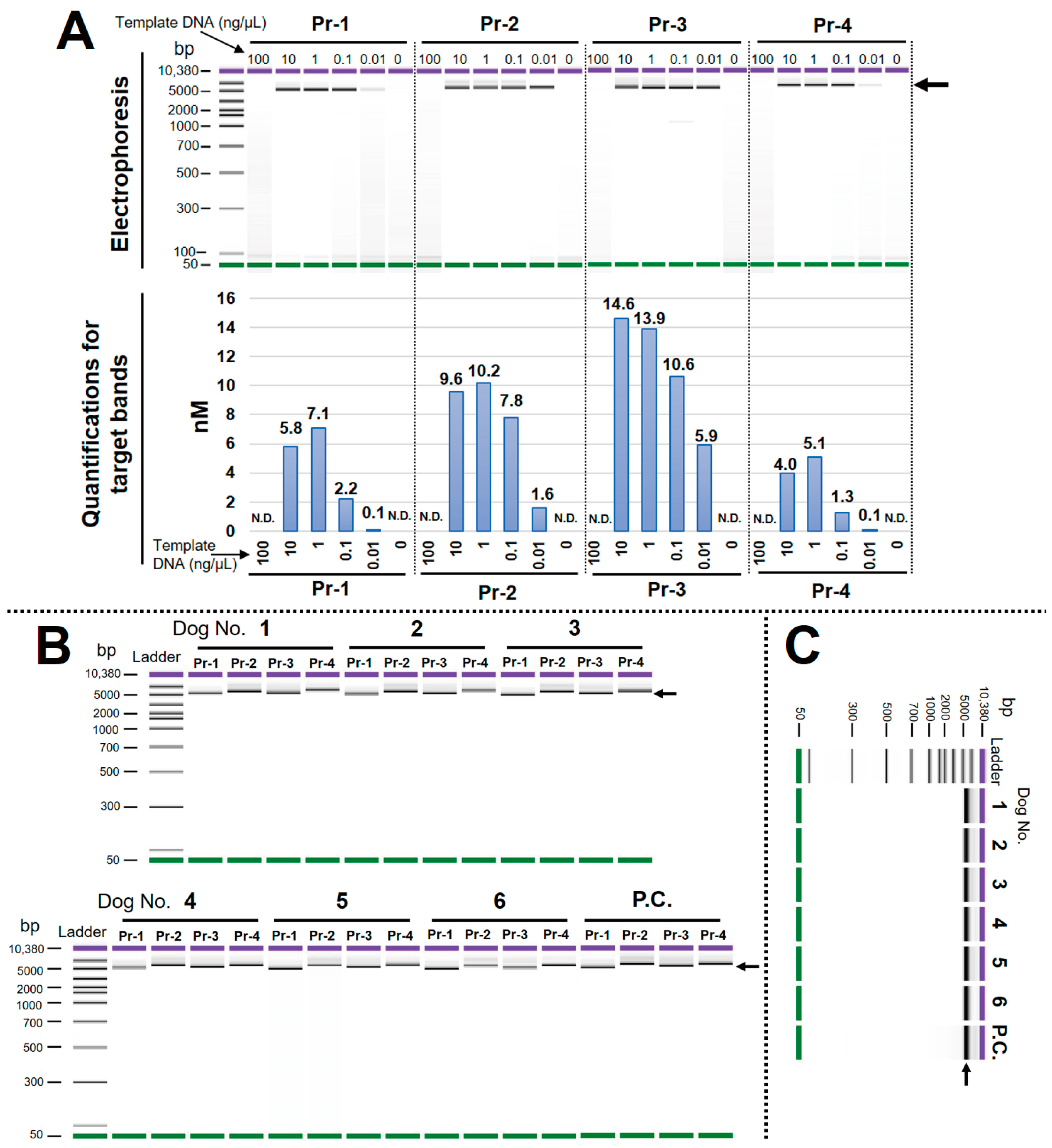
3.2. Optimal LR-PCR Condition Was Determined, and the High Specificity of Each Primer Pair Was Confirmed
3.3. Whole Mitochondrial DNA Was Accurately Resequenced in All Dogs
3.4. Phylogenetic Tree Analysis Confirmed the Similarity among Dogs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, L.H. Mitochondrial DNA. Available online: https://www.genome.gov/genetics-glossary/Mitochondrial-DNA (accessed on 23 February 2023).
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.-C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Hudson, G. Mitochondrial Genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef]
- Taanman, J.-W. The Mitochondrial Genome: Structure, Transcription, Translation and Replication. Biochim. Biophys. Acta Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Homo Sapiens Mitochondrion, Complete Genome—Nucleotide—NCBI. Available online: https://www.ncbi.nlm.nih.gov/nuccore/NC_012920.1/ (accessed on 23 February 2023).
- Canis lupus familiaris Mitochondrion, Complete Genome—Nucleotide—NCBI. Available online: https://www.ncbi.nlm.nih.gov/nuccore/NC_002008.4 (accessed on 23 February 2023).
- Shinoda, K.-I. Analysis of DNA Variations Reveals the Origins and Dispersal of Modern Humans. J. Geogr. 2009, 118, 311–319. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and Organization of the Human Mitochondrial Genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Lyons, E.A.; Scheible, M.K.; Sturk-Andreaggi, K.; Irwin, J.A.; Just, R.S. A High-Throughput Sanger Strategy for Human Mitochondrial Genome Sequencing. BMC Genom. 2013, 14, 881. [Google Scholar] [CrossRef]
- Yao, Y.; Nishimura, M.; Murayama, K.; Kuranobu, N.; Tojo, S.; Beppu, M.; Ishige, T.; Itoga, S.; Tsuchida, S.; Mori, M.; et al. A Simple Method for Sequencing the Whole Human Mitochondrial Genome Directly from Samples and Its Application to Genetic Testing. Sci. Rep. 2019, 9, 17411. [Google Scholar] [CrossRef]
- Zhou, K.; Liu, Y.; Yuan, Q.; Lai, D.; Guo, S.; Wang, Z.; Su, L.; Zhang, H.; Wang, X.; Guo, W.; et al. Next-Generation Sequencing-Based Analysis of Urine Cell-Free MtDNA Reveals Aberrant Fragmentation and Mutation Profile in Cancer Patients. Clin. Chem. 2022, 68, 561–573. [Google Scholar] [CrossRef]
- Ivanova, E.M.; Kandilarova, S.M.; Lukanov, T.I.; Naumova, E.J.; Akabalieva, K.V.; Milanova, V.K. NGS-Based MtDNA Profiling Could Reveal Genetic Alterations in Schizophrenia. Curr. Top. Med. Chem. 2021, 21, 938–948. [Google Scholar] [CrossRef]
- Yin, C.; Li, D.Y.; Guo, X.; Cao, H.Y.; Chen, Y.B.; Zhou, F.; Ge, N.J.; Liu, Y.; Guo, S.S.; Zhao, Z.; et al. NGS-Based Profiling Reveals a Critical Contributing Role of Somatic D-Loop MtDNA Mutations in HBV-Related Hepatocarcinogenesis. Ann. Oncol. 2019, 30, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Balciuniene, J.; Diaz-Miranda, M.A.; McCormick, E.M.; Aref-Eshghi, E.; Muir, A.M.; Cao, K.; Troiani, J.; Moseley, A.; Fan, Z.; et al. Advanced Approach for Comprehensive MtDNA Genome Testing in Mitochondrial Disease. Mol. Genet. Metab. 2022, 135, 93–101. [Google Scholar] [CrossRef]
- Mahmud, S.; Biswas, S.; Afrose, S.; Mita, M.A.; Hasan, M.R.; Shimu, M.S.S.; Paul, G.K.; Chung, S.; Saleh, M.A.; Alshehri, S.; et al. Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders. Curr. Issues Mol. Biol. 2022, 44, 1127–1148. [Google Scholar] [CrossRef]
- Kivisild, T. Maternal Ancestry and Population History from Whole Mitochondrial Genomes. Investig. Genet. 2015, 6, 3. [Google Scholar] [CrossRef]
- Key Indicator Summary, Survey of Dog and Cat Ownership in Japan, Japan Pet Food Association. Available online: https://petfood.or.jp/data/chart2022/index.html (accessed on 23 February 2023).
- Tkaczyk-Wlizło, A.; Kowal, K.; Ślaska, B. Mitochondrial DNA Alterations in the Domestic Dog (Canis lupus familiaris) and Their Association with Development of Diseases: A Review. Mitochondrion 2022, 63, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S. A Review of Mitochondrial Disease in Dogs. Companion Anim. 2021, 26, 257–264. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Canis lupus familiaris Genome Assembly Dog10K_Boxer_Tasha. Available online: https://www.ncbi.nlm.nih.gov/data-hub/genome/GCF_000002285.5/ (accessed on 1 March 2023).
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2020, 48, D84–D86. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kowal, K.; Ślaska, B.; Bownik, A.; Horecka, B.; Gawor, J.; Śmiech, A.; Tkaczyk, A. Analysis of Mitochondrial Genome from Labrador (Canis lupus familiaris) with Mammary Gland Tumour Reveals Novel Mutations and Polymorphisms. Ann. Anim. Sci. 2019, 19, 619–632. [Google Scholar] [CrossRef]
- Kowal, K.; Tkaczyk-Wlizło, A.; Pierzchała, M.; Gawor, J.; Ślaska, B. Molecular Differences in Mitochondrial DNA Genomes of Dogs with Malignant Mammary Tumours. Vet. Comp. Oncol. 2022, 20, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Hsiou, C.-L.; Hsu, C.-C.; Liao, P.-W.; Yang, F.-H.; Lee, A.N.; Huang, W.-H. Forensic Death Investigations of Dog Bite Injuries in 31 Cats. Animals 2022, 12, 2404. [Google Scholar] [CrossRef]
- Rahman, M.M.; Ali, M.E.; Hamid, S.B.A.; Mustafa, S.; Hashim, U.; Hanapi, U.K. Polymerase Chain Reaction Assay Targeting Cytochrome b Gene for the Detection of Dog Meat Adulteration in Meatball Formulation. Meat Sci. 2014, 97, 404–409. [Google Scholar] [CrossRef]
- Barrientos, L.S.; Crespi, J.A.; Fameli, A.; Posik, D.M.; Morales, H.; Peral García, P.; Giovambattista, G. DNA Profile of Dog Feces as Evidence to Solve a Homicide. Leg. Med. 2016, 22, 54–57. [Google Scholar] [CrossRef]
- Linacre, A. Animal Forensic Genetics. Genes 2021, 12, 515. [Google Scholar] [CrossRef]
- Statistical Material “Situation of Collection of Dogs and Cats, Accommodation and Disposal of Injured Animals, etc.”, Welfare and Appropriate Management of Animals, Ministry of the Environment, Government of Japan. Available online: https://www.env.go.jp/nature/dobutsu/aigo/2_data/statistics/dog-cat.html (accessed on 11 June 2023).
- About Microchip Information Registration for Dogs and Cats, Welfare and Appropriate Management of Animals, Ministry of the Environment, Government of Japan. Available online: https://www.env.go.jp/nature/dobutsu/aigo/pickup/chip.html (accessed on 11 June 2023).
- Yachie, N.; Robotic Biology Consortium; Natsume, T. Robotic Crowd Biology with Maholo LabDroids. Nat. Biotechnol. 2017, 35, 310–312. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dog No. | Dog Breed/Gender | Total Length (bp) of mtDNA | Number of Variants | Coverage of Variants | Number of Non-Synonymous Amino Acids | Pathogenic Variant | ||
|---|---|---|---|---|---|---|---|---|
| 1 | Kooikerhondje/female | 16,730 | SNV | 101 | Total: 104 | 6135 to 51,966 (Average: 20,178) | 17 | N/A |
| MNV | 1 | |||||||
| Insertion | 2 | |||||||
| Deletion | 0 | |||||||
| 2 | Keeshond/ female | 16,732 | SNV | 33 | Total: 36 | 6525 to 24,492 (Average: 13,442) | 4 | N/A |
| MNV | 0 | |||||||
| Insertion | 3 | |||||||
| Deletion | 0 | |||||||
| 3 | Keeshond/ female | 16,732 | SNV | 33 | Total: 36 | 7978 to 33,320 (Average: 17,579) | 4 | N/A |
| MNV | 0 | |||||||
| Insertion | 3 | |||||||
| Deletion | 0 | |||||||
| 4 | Shiba Inu/ female | 16,730 | SNV | 105 | Total: 109 | 6300 to 45,580 (Average: 17,999) | 20 | N/A |
| MNV | 0 | |||||||
| Insertion | 3 | |||||||
| Deletion | 1 | |||||||
| 5 | Shiba Inu/ male | 16,730 | SNV | 23 | Total: 25 | 12,637 to 36,862 (Average: 21,158) | 3 | N/A |
| MNV | 0 | |||||||
| Insertion | 2 | |||||||
| Deletion | 0 | |||||||
| 6 | Beagle/ male | 16,730 | SNV | 21 | Total: 23 | 9690 to 44,299 (Average: 22,209) | 3 | N/A |
| MNV | 0 | |||||||
| Insertion | 2 | |||||||
| Deletion | 0 | |||||||
| P.C. | Toy Poodle/ male | 16,730 | SNV | 27 | Total: 29 | 9122 to 28,966 (Average: 17,035) | 3 | N/A |
| MNV | 0 | |||||||
| Insertion | 2 | |||||||
| Deletion | 0 | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugasawa, T.; Matsumoto, Y.; Fang, H.; Takemasa, T.; Komine, R.; Tamai, S.; Gu, W.; Tanaka, K.; Kanki, Y.; Takahashi, Y. Establishing a Sequencing Method for the Whole Mitochondrial DNA of Domestic Dogs. Animals 2023, 13, 2332. https://doi.org/10.3390/ani13142332
Sugasawa T, Matsumoto Y, Fang H, Takemasa T, Komine R, Tamai S, Gu W, Tanaka K, Kanki Y, Takahashi Y. Establishing a Sequencing Method for the Whole Mitochondrial DNA of Domestic Dogs. Animals. 2023; 13(14):2332. https://doi.org/10.3390/ani13142332
Chicago/Turabian StyleSugasawa, Takehito, Yuki Matsumoto, Hui Fang, Tohru Takemasa, Ritsuko Komine, Shinsuke Tamai, Wenchao Gu, Kei Tanaka, Yasuharu Kanki, and Yoichiro Takahashi. 2023. "Establishing a Sequencing Method for the Whole Mitochondrial DNA of Domestic Dogs" Animals 13, no. 14: 2332. https://doi.org/10.3390/ani13142332
APA StyleSugasawa, T., Matsumoto, Y., Fang, H., Takemasa, T., Komine, R., Tamai, S., Gu, W., Tanaka, K., Kanki, Y., & Takahashi, Y. (2023). Establishing a Sequencing Method for the Whole Mitochondrial DNA of Domestic Dogs. Animals, 13(14), 2332. https://doi.org/10.3390/ani13142332