
Genomic Characterization and Initial Insight into Mastitis-Associated SNP Profiles of Local Latvian Bos taurus Breeds
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Animals and Sample Collection
2.2. Milk Sample Collection and Bacteriological Testing
2.3. DNA Extraction
2.4. Library Construction and Sequencing Analysis
2.5. Sequencing Data Processing and Variant Calling
2.6. Genomic Analysis
2.7. Population Structure
2.8. Detection of Variants Associated with Mastitis
2.9. Statistical Analysis of SCC Data
3. Results
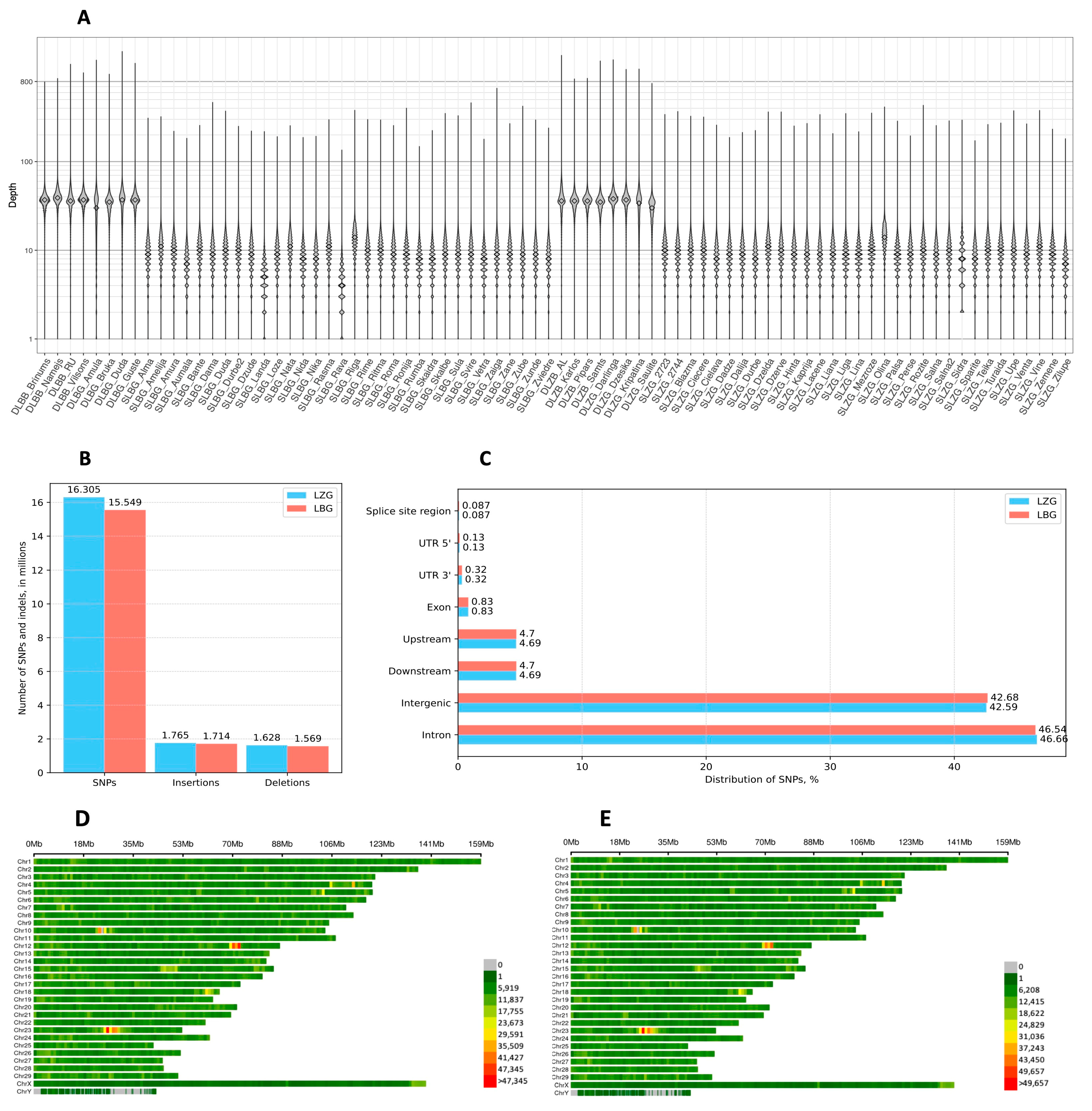
3.1. Variant Calling and Distribution of Polymorphisms
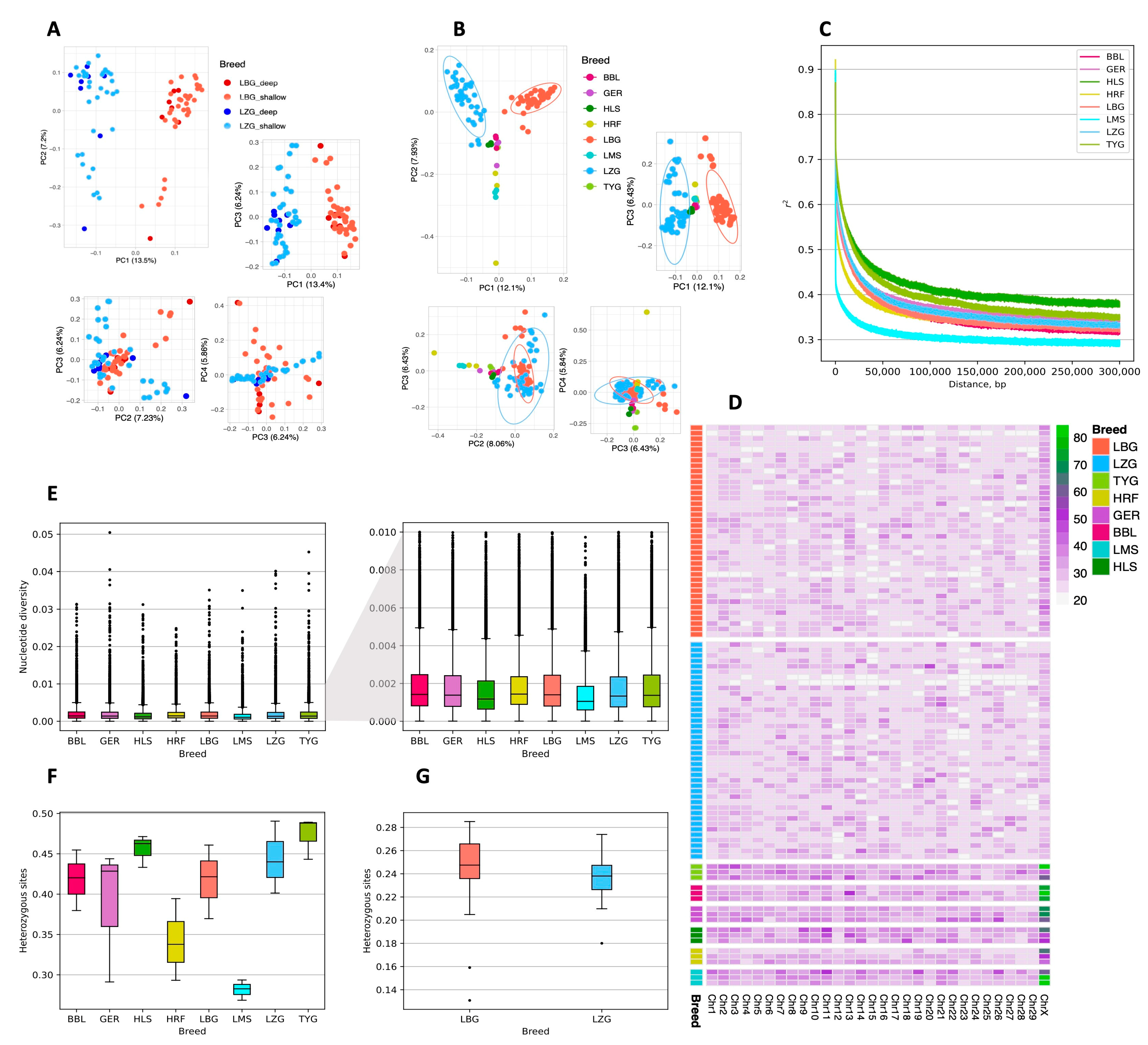
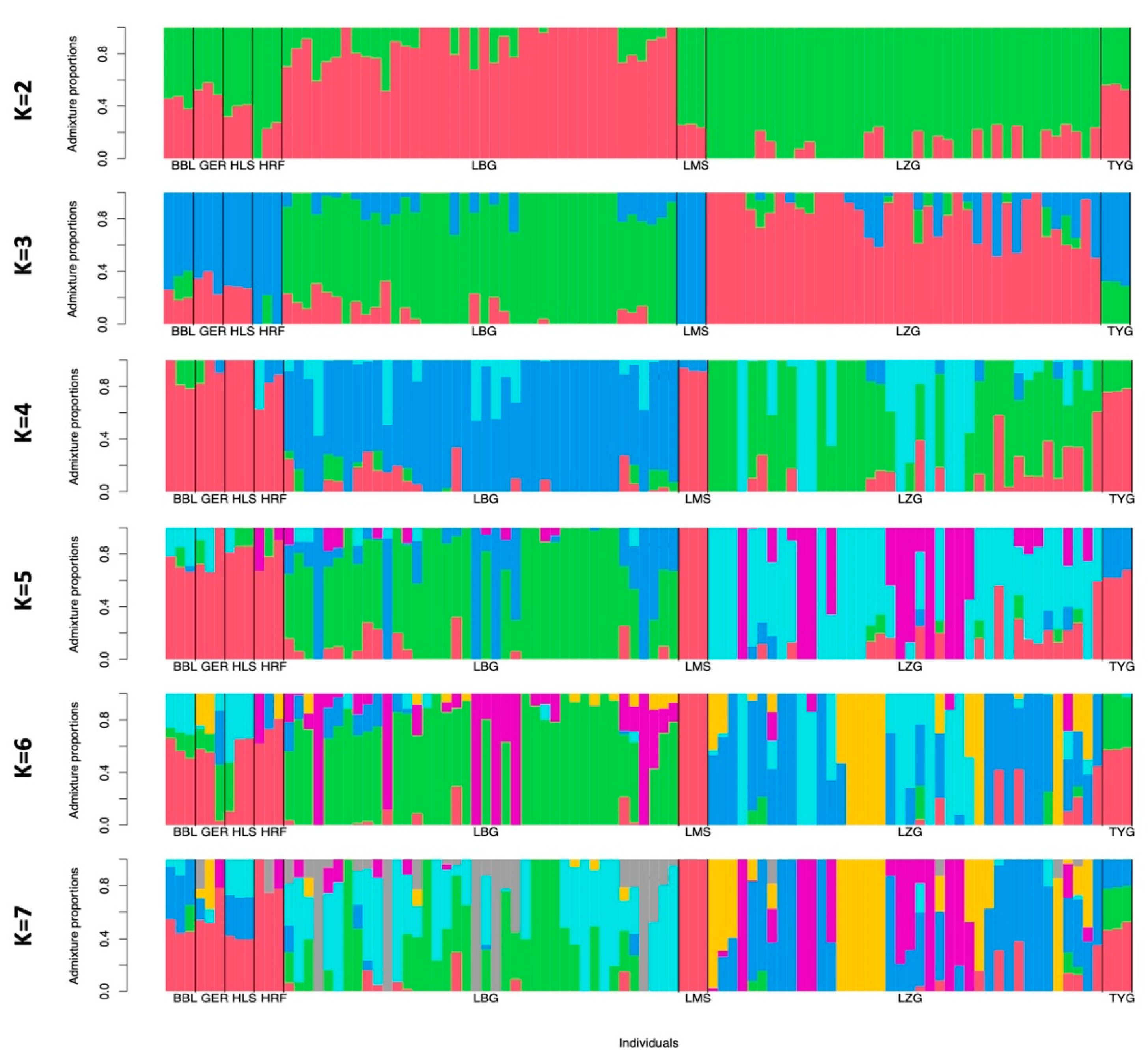
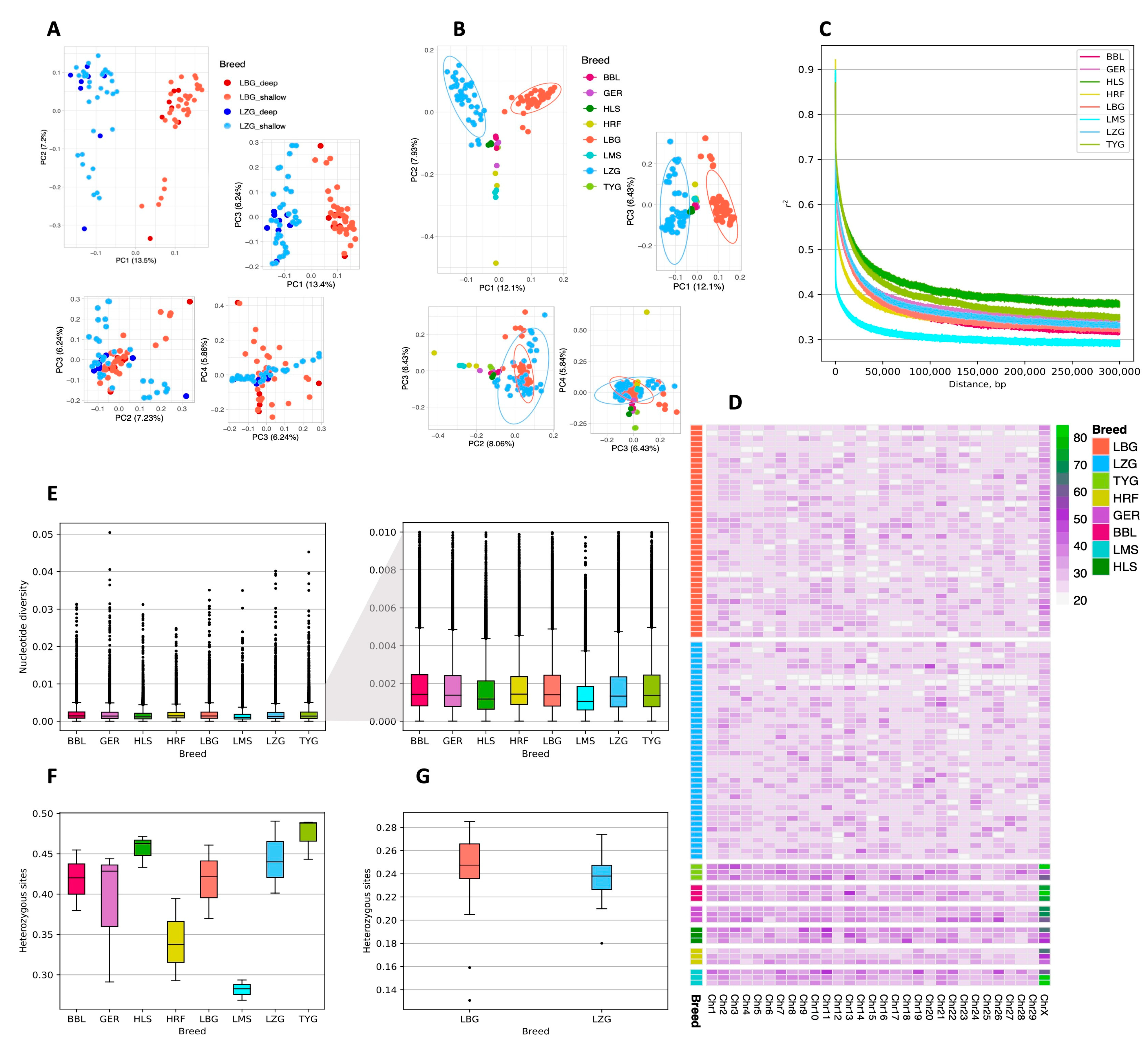
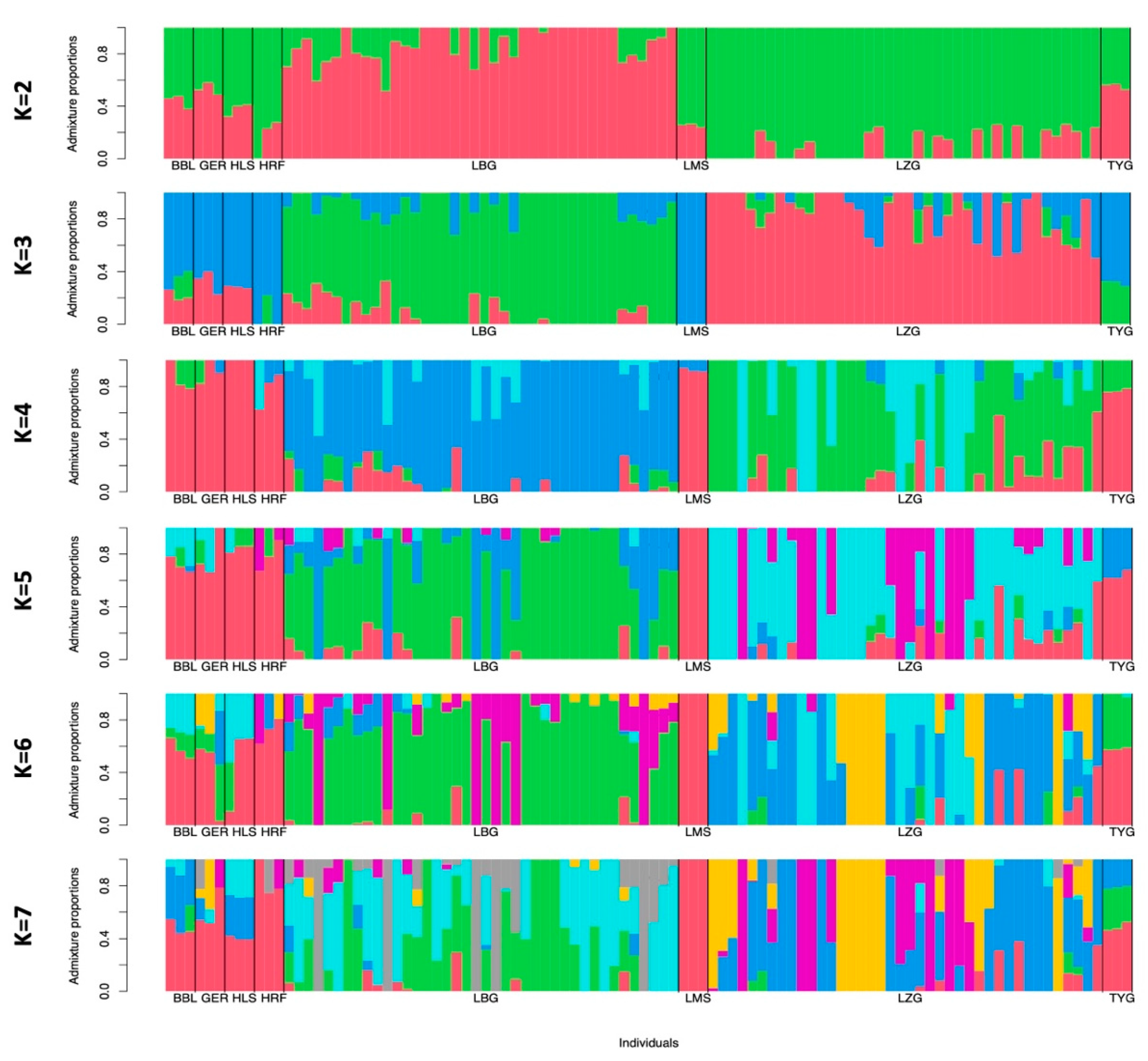
3.2. Population Genetics
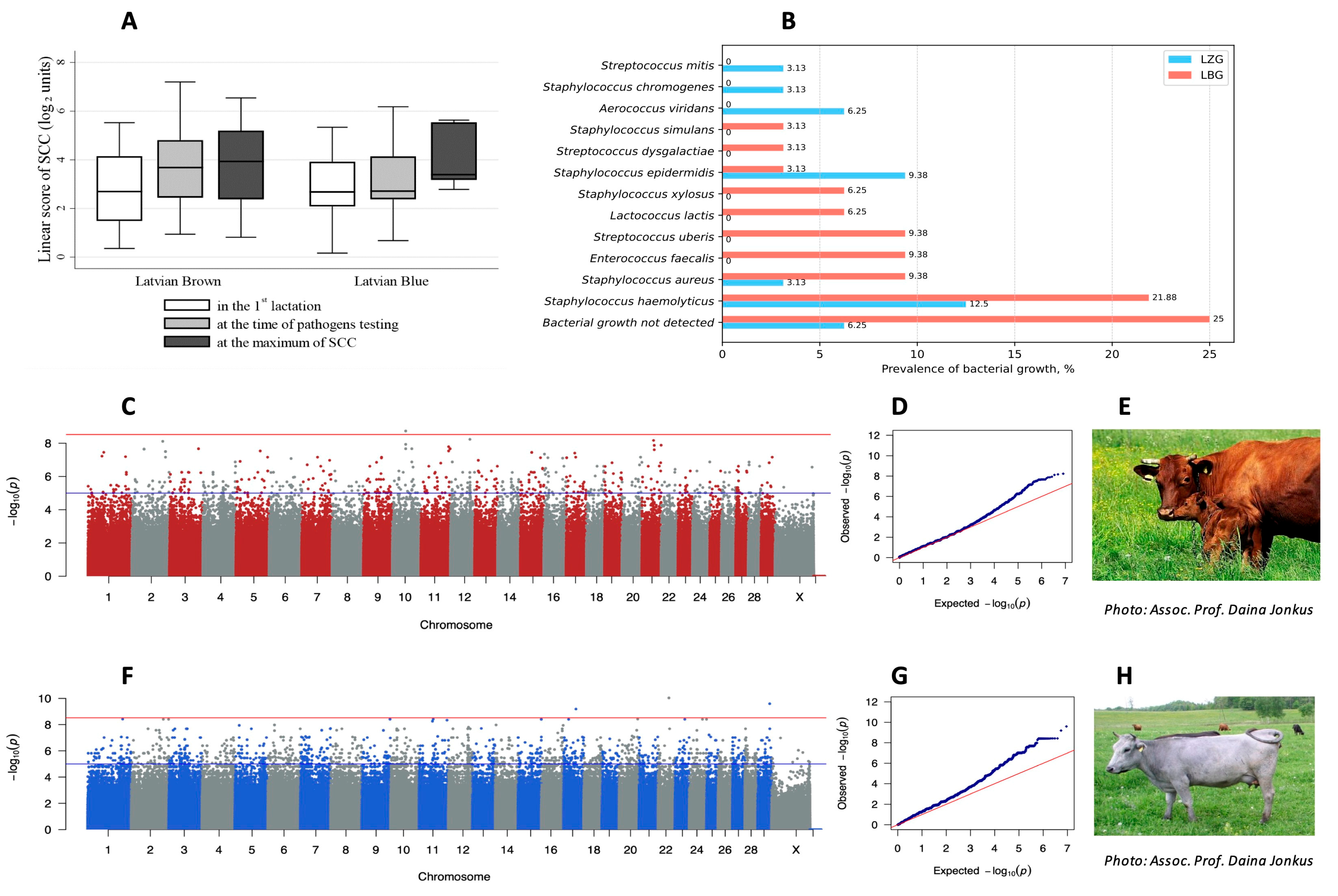
3.3. Variant Association with Mastitis
4. Discussion
4.1. Genetic Structure of Breeds
4.2. Mastitis Resistance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United Nations Environment Programme CBD Handbook. In Convention on Biological Diversity; 1992; pp. 1–28. Available online: https://www.cbd.int/doc/legal/cbd-en.pdf (accessed on 4 July 2023).
- Morgan-Davies, J.; Morgan-Davies, C.; Pollock, M.L.; Holland, J.P.; Waterhouse, A. Characterisation of Extensive Beef Cattle Systems: Disparities between Opinions, Practice and Policy. Land Use Policy 2014, 38, 707–718. [Google Scholar] [CrossRef]
- Regulation (EU) 2016/1012 of the European Parliament and of the Council of 8 June 2016 Clause 24 of Article 2. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32016R1012&qid=1689913000067 (accessed on 24 July 2023).
- List of Breed Societies and Breeding Operations in Latvia. Available online: https://www.ldc.gov.lv/lv/media/663/download?attachment (accessed on 24 July 2023).
- Latvian Brown Cow’s Conservation Program from 2019 and on (In Latvian). Available online: https://www.ldc.gov.lv/lv/media/93/download?attachment (accessed on 24 July 2023).
- Breeding Program of the Latvian Blue Cow Breed 2019–2029 (In Latvian). Available online: google.com/search?q=latvijas+zilāds+škirnes+audzēšanas+programma&rlz=1C1GCEA_enLV785LV785&oq=latvijas+zilāds+škirnes+audzēšanas+programma&gs_lcrp=EgZjaHJvbWUyBggAEEUYOTIJCAEQIRgKGKAB0gENMTc4MjMzNzZqMGoxNagCALACAA&sourceid=chrome&ie=UTF-8 (accessed on 24 July 2023).
- European Regional Focal Point for Animal Genetic Resources (ERFP). Available online: https://www.animalgeneticresources.net/index.php/country/latvia/#1589182530764-d1c43160-c850 (accessed on 4 July 2023).
- Dmitriev, N.G.; Ernst, L.K. Animal Genetic Resources of the USSR. In Animal Production and Health Paper; FAO: Rome, Italy, 1989; pp. 1–517. [Google Scholar]
- Agricultural Data Centre Public Register Information. Available online: https://registri.ldc.gov.lv/en/?lang=en (accessed on 2 May 2022).
- Consultative Committee of Farm Animal Genetic Resources. State of the Animal Genetic Resources of Latvia; Ministry of Agriculture of Latvia, Rīga, Latvia. 2003. Available online: https://www.fao.org/3/a1250e/annexes/CountryReports/Latvia.pdf (accessed on 4 July 2023).
- Curone, G.; Filipe, J.; Cremonesi, P.; Trevisi, E.; Amadori, M.; Pollera, C.; Castiglioni, B.; Turin, L.; Tedde, V.; Vigo, D.; et al. What We Have Lost: Mastitis Resistance in Holstein Friesians and in a Local Cattle Breed. Res. Vet. Sci. 2018, 116, 88–98. [Google Scholar] [CrossRef]
- Trujano-Chavez, M.Z.; Sánchez-Ramos, R.; Pérez-Rodríguez, P.; Ruíz-Flores, A. Genetic Diversity and Population Structure for Resistance and Susceptibility to Mastitis in Braunvieh Cattle. Vet. Sci. 2021, 8, 329. [Google Scholar] [CrossRef]
- Cheng, W.N.; Han, S.G. Bovine Mastitis: Risk Factors, Therapeutic Strategies, and Alternative Treatments—A Review. Asian-Australas. J. Anim. Sci. 2020, 33, 1699–1713. [Google Scholar] [CrossRef]
- Krishnamoorthy, P.; Goudar, A.L.; Suresh, K.P.; Roy, P. Global and Countrywide Prevalence of Subclinical and Clinical Mastitis in Dairy Cattle and Buffaloes by Systematic Review and Meta-Analysis. Res. Vet. Sci. 2021, 136, 561–586. [Google Scholar] [CrossRef]
- Haxhiaj, K.; Wishart, D.S.; Ametaj, B.N. Mastitis: What It Is, Current Diagnostics, and the Potential of Metabolomics to Identify New Predictive Biomarkers. Dairy 2022, 3, 722–746. [Google Scholar] [CrossRef]
- Zigo, F.; Farkašová, Z.; Výrostková, J.; Regecová, I.; Ondrašovičová, S.; Vargová, M.; Sasáková, N.; Pecka-Kielb, E.; Bursová, Š.; Kiss, D.S. Dairy Cows’ Udder Pathogens and Occurrence of Virulence Factors in Staphylococci. Animals 2022, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- De Vliegher, S.; Fox, L.K.; Piepers, S.; McDougall, S.; Barkema, H.W. Invited Review: Mastitis in Dairy Heifers: Nature of the Disease, Potential Impact, Prevention, and Control. J. Dairy Sci. 2012, 95, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Doherr, M.G.; Roesch, M.; Schaeren, W.; Schallibaum, M.; Blum, J.W. Risk Factors Associated with Subclinical Mastitis in Dairy Cows on Swiss Organic and Conventional Production System Farms. Veterinární Medicína 2007, 52, 487–495. [Google Scholar] [CrossRef]
- Narayana, S.G.; de Jong, E.; Schenkel, F.S.; Fonseca, P.A.S.; Chud, T.C.S.; Powell, D.; Wachoski-Dark, G.; Ronksley, P.E.; Miglior, F.; Orsel, K.; et al. Underlying Genetic Architecture of Resistance to Mastitis in Dairy Cattle: A Systematic Review and Gene Prioritization Analysis of Genome-Wide Association Studies. J. Dairy Sci. 2023, 106, 323–351. [Google Scholar] [CrossRef]
- Brajnik, Z.; Ogorevc, J. Candidate Genes for Mastitis Resistance in Dairy Cattle: A Data Integration Approach. J. Anim. Sci. Biotechnol. 2023, 14, 10. [Google Scholar] [CrossRef]
- Bakhtiarizadeh, M.R.; Mirzaei, S.; Norouzi, M.; Sheybani, N.; Vafaei Sadi, M.S. Identification of Gene Modules and Hub Genes Involved in Mastitis Development Using a Systems Biology Approach. Front. Genet. 2020, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, C.; Stålhammar, H.; Strandberg, E.; Eriksson, S.; Fikse, W.F. Association of Genomically Enhanced and Parent Average Breeding Values with Cow Performance in Nordic Dairy Cattle. J. Dairy Sci. 2020, 103, 6383–6391. [Google Scholar] [CrossRef]
- Lu, X.; Jiang, H.; Arbab, A.A.I.; Wang, B.; Liu, D.; Abdalla, I.M.; Xu, T.; Sun, Y.; Liu, Z.; Yang, Z. Investigating Genetic Characteristics of Chinese Holstein Cow’s Milk Somatic Cell Score by Genetic Parameter Estimation and Genome-Wide Association. Agriculture 2023, 13, 267. [Google Scholar] [CrossRef]
- Bijma, P.; Hulst, A.D.; de Jong, M.C.M. The Quantitative Genetics of the Prevalence of Infectious Diseases: Hidden Genetic Variation Due to Indirect Genetic Effects Dominates Heritable Variation and Response to Selection. Genetics 2022, 220, 141. [Google Scholar] [CrossRef]
- Weigel, K.A.; Shook, G.E. Genetic Selection for Mastitis Resistance. Vet. Clin. N. Am. Food Anim. Pract. 2018, 34, 457–472. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the International AAAI Conference on Web and Social Media, San Jose, CA, USA, 17–20 May 2009; Volume 3, pp. 361–362. [Google Scholar] [CrossRef]
- Firdaus, I. Laboratory Handbook on Bovine Mastitis, 3rd ed.; National Mastitis Council (NMC): New Prague, MN, USA, 2017. [Google Scholar]
- Rovite, V.; Wolff-Sagi, Y.; Zaharenko, L.; Nikitina-Zake, L.; Grens, E.; Klovins, J. Genome Database of the Latvian Population (LGDB): Design, Goals, and Primary Results. J. Epidemiol. 2018, 28, 353–360. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Hayes, B.J.; Daetwyler, H.D. 1000 Bull Genomes Project to Map Simple and Complex Genetic Traits in Cattle: Applications and Outcomes. Annu. Rev. Anim. Biosci. 2019, 7, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Geraldine, A.; der Auwera, V.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2020; ISBN 9781491975190. [Google Scholar]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. bioRxiv 2011. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. RMVP: A Memory-Efficient, Visualization-Enhanced, and Parallel-Accelerated Tool for Genome-Wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr : An R Package for Genetic Analysis of Populations with Clonal, Partially Clonal, and/or Sexual Reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed]
- Turner, D. Qqman: An R Package for Visualizing GWAS Results Using Q-Q and Manhattan Plots. J. Open Source Softw. 2018, 3, 731. [Google Scholar] [CrossRef]
- Czech, B.; Guldbrandtsen, B.; Szyda, J. Patterns of DNA Variation between the Autosomes, the X Chromosome and the Y Chromosome in Bos Taurus Genome. Sci. Rep. 2020, 10, 13641. [Google Scholar] [CrossRef]
- Neumann, G.B.; Korkuć, P.; Arends, D.; Wolf, M.J.; May, K.; König, S.; Brockmann, G.A. Genomic Diversity and Relationship Analyses of Endangered German Black Pied Cattle (DSN) to 68 Other Taurine Breeds Based on Whole-Genome Sequencing. Front. Genet. 2023, 13, 993959. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-H.; Kantanen, J. Genetic Structure of Eurasian Cattle (Bos Taurus) Based on Microsatellites: Clarification for Their Breed Classification. Anim. Genet. 2010, 41, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-H.; Kantanen, J.; Michelson, A.; Saarma, U. Genetic Components of Grey Cattle in Estonia as Revealed by Microsatellite Analysis Using Two Bayesian Clustering Methods. BMC Res. Notes 2011, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Doublet, A.-C.; Croiseau, P.; Fritz, S.; Michenet, A.; Hozé, C.; Danchin-Burge, C.; Laloë, D.; Restoux, G. The Impact of Genomic Selection on Genetic Diversity and Genetic Gain in Three French Dairy Cattle Breeds. Genet. Sel. Evol. 2019, 51, 52. [Google Scholar] [CrossRef]
- Bhati, M.; Kadri, N.K.; Crysnanto, D.; Pausch, H. Assessing Genomic Diversity and Signatures of Selection in Original Braunvieh Cattle Using Whole-Genome Sequencing Data. BMC Genom. 2020, 21, 27. [Google Scholar] [CrossRef]
- Weldenegodguad, M.; Popov, R.; Pokharel, K.; Ammosov, I.; Ming, Y.; Ivanova, Z.; Kantanen, J. Whole-Genome Sequencing of Three Native Cattle Breeds Originating From the Northernmost Cattle Farming Regions. Front. Genet. 2019, 9, 728. [Google Scholar] [CrossRef]
- Senczuk, G.; Mastrangelo, S.; Ciani, E.; Battaglini, L.; Cendron, F.; Ciampolini, R.; Crepaldi, P.; Mantovani, R.; Bongioni, G.; Pagnacco, G.; et al. The Genetic Heritage of Alpine Local Cattle Breeds Using Genomic SNP Data. Genet. Sel. Evol. 2020, 52, 40. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Ciani, E.; Ajmone Marsan, P.; Bagnato, A.; Battaglini, L.; Bozzi, R.; Carta, A.; Catillo, G.; Cassandro, M.; Casu, S.; et al. Conservation Status and Historical Relatedness of Italian Cattle Breeds. Genet. Sel. Evol. 2018, 50, 35. [Google Scholar] [CrossRef]
- Schmidtmann, C.; Schönherz, A.; Guldbrandtsen, B.; Marjanovic, J.; Calus, M.; Hinrichs, D.; Thaller, G. Assessing the Genetic Background and Genomic Relatedness of Red Cattle Populations Originating from Northern Europe. Genet. Sel. Evol. 2021, 53, 23. [Google Scholar] [CrossRef]
- Benjelloun, B.; Boyer, F.; Streeter, I.; Zamani, W.; Engelen, S.; Alberti, A.; Alberto, F.J.; BenBati, M.; Ibnelbachyr, M.; Chentouf, M.; et al. An Evaluation of Sequencing Coverage and Genotyping Strategies to Assess Neutral and Adaptive Diversity. Mol. Ecol. Resour. 2019, 19, 1497–1515. [Google Scholar] [CrossRef]
- Qanbari, S. On the Extent of Linkage Disequilibrium in the Genome of Farm Animals. Front. Genet. 2020, 10, 1304. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.C.; Dadousis, C.; Bozzi, R. Estimation of Linkage Disequilibrium and Effective Population Size in Three Italian Autochthonous Beef Breeds. Animals 2020, 10, 1034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Calus, M.P.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Estimation of Inbreeding Using Pedigree, 50k SNP Chip Genotypes and Full Sequence Data in Three Cattle Breeds. BMC Genet. 2015, 16, 88. [Google Scholar] [CrossRef] [PubMed]
- Viinalass, H.; Värv, S.; Boveiniene, B.; Bekere, R. Characterisation of Cattle Breeds in Baltic Countries by Genetic Markers. Biologija 2002, 48, 16–19. [Google Scholar]
- Paura, L.; Jonkus, D. Inbreeding Evaluation in Latvian Local Cattle Breeds. Acta Fytotech. Et Zootech. 2020, 23, 52–57. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.C.d.C.; Ventura, R.V.; Baldi, F.; da Silva, M.V.G.B. Assessment of Runs of Homozygosity Islands and Estimates of Genomic Inbreeding in Gyr (Bos Indicus) Dairy Cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef]
- Al-Mamun, H.A.; A Clark, S.; Kwan, P.; Gondro, C. Genome-Wide Linkage Disequilibrium and Genetic Diversity in Five Populations of Australian Domestic Sheep. Genet. Sel. Evol. 2015, 47, 90. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.-F.; Chen, Q.-M.; Zhang, F.-W.; Hanif, Q.; Huang, B.-Z.; Chen, N.-B.; Qu, K.-X.; Zhan, J.-X.; Chen, H.; Jiang, Y.; et al. Genome-wide association study identifies quantitative trait loci affecting cattle temperament. Zool. Res. 2022, 43, 14–25. [Google Scholar] [CrossRef]
- Korec, E.; Ungrová, L.; Hejnar, J.; Grieblová, A.; Zelená, K. Three new genes associated with longevity in the European Bison. Vet. Anim. Sci. 2022, 17, 100266. [Google Scholar] [CrossRef] [PubMed]
- Nanaei, H.A.; Qanatqestani, M.D.; Esmailizadeh, A. Whole-genome resequencing reveals selection signatures associated with milk production traits in African Kenana dairy zebu cattle. Genomics 2020, 112, 880–885. [Google Scholar] [CrossRef]
- Tijjani, A.; Utsunomiya, Y.T.; Ezekwe, A.G.; Nashiru, O.; Hanotte, O. Genome Sequence Analysis Reveals Selection Signatures in Endangered Trypanotolerant West African Muturu Cattle. Front. Genet. 2019, 10, 442. [Google Scholar] [CrossRef]
- Chen, Q.; Xu l Zhang, M.; Zhang, T.; Yan, M.; Zhai, M.; Huang, X. Whole genome resequencing reveals the genetic contribution of Kazakh and Swiss Brown cattle to a population of Xinjiang Brown cattle. Gene 2022, 839, 146725. [Google Scholar] [CrossRef]
- Chhotaray, S.; Panigrahi, M.; Bhushan, B.; Gaur, G.K.; Dutt, T.; Mishra, B.P.; Singh, R.K. Genome-wide association study reveals genes crucial for coat color production in Vrindavani cattle. Livest. Sci. 2021, 247, 104476. [Google Scholar] [CrossRef]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-Wide Association Studies. Nat. Rev. Methods Primers 2021, 1, 59. [Google Scholar] [CrossRef]
- Kuo, T.; Kraakman, M.J.; Damle, M.; Gill, R.; Lazar, M.A.; Accili, D. Identification of C2CD4A as a Human Diabetes Susceptibility Gene with a Role in β Cell Insulin Secretion. Proc. Natl. Acad. Sci. USA 2019, 116, 20033–20042. [Google Scholar] [CrossRef]
- Warton, K.; Foster, N.C.; Gold, W.A.; Stanley, K.K. A Novel Gene Family Induced by Acute Inflammation in Endothelial Cells. Gene 2004, 342, 85–95. [Google Scholar] [CrossRef]
- Brand, B.; Hartmann, A.; Repsilber, D.; Griesbeck-Zilch, B.; Wellnitz, O.; Kühn, C.; Ponsuksili, S.; Meyer, H.H.; Schwerin, M. Comparative Expression Profiling of E. Coli and S. Aureus Inoculated Primary Mammary Gland Cells Sampled from Cows with Different Genetic Predispositions for Somatic Cell Score. Genet. Sel. Evol. 2011, 43, 24. [Google Scholar] [CrossRef]
- Oliveira, H.R.; Lourenco, D.A.L.; Masuda, Y.; Misztal, I.; Tsuruta, S.; Jamrozik, J.; Brito, L.F.; Silva, F.F.; Cant, J.P.; Schenkel, F.S. Single-Step Genome-Wide Association for Longitudinal Traits of Canadian Ayrshire, Holstein, and Jersey Dairy Cattle. J. Dairy Sci. 2019, 102, 9995–10011. [Google Scholar] [CrossRef] [PubMed]
- Stoeckle, C.; Gouttefangeas, C.; Hammer, M.; Weber, E.; Melms, A.; Tolosa, E. Cathepsin W Expressed Exclusively in CD8+ T Cells and NK Cells, Is Secreted during Target Cell Killing but Is Not Essential for Cytotoxicity in Human CTLs. Exp. Hematol. 2009, 37, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Younis, S.; Javed, Q.; Blumenberg, M. Meta-Analysis of Transcriptional Responses to Mastitis-Causing Escherichia Coli. PLoS ONE 2016, 11, e0148562. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR Signaling in Health and Disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Günther, J.; Petzl, W.; Bauer, I.; Ponsuksili, S.; Zerbe, H.; Schuberth, H.-J.; Brunner, R.M.; Seyfert, H.-M. Differentiating Staphylococcus Aureus from Escherichia Coli Mastitis: S. Aureus Triggers Unbalanced Immune-Dampening and Host Cell Invasion Immediately after Udder Infection. Sci. Rep. 2017, 7, 4811. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.G.; Miller, R.K.; Nicholson, K.L.; Gill, C.A.; Herring, A.D.; Riggs, P.K.; Sawyer, J.E.; Savell, J.W.; Sanders, J.O. Genome Association of Carcass and Palatability Traits from Bos Indicus-Bos Taurus Crossbred Steers within Electrical Stimulation Status and Correspondence with Steer Temperament 2. Palatability. Livest. Sci. 2020, 232, 103897. [Google Scholar] [CrossRef]
- Iwai, A.; Hijikata, M.; Hishiki, T.; Isono, O.; Chiba, T.; Shimotohno, K. Coiled-Coil Domain Containing 85B Suppresses the β-Catenin Activity in a P53-Dependent Manner. Oncogene 2008, 27, 1520–1526. [Google Scholar] [CrossRef]
- Abdalla, I.M.; Lu, X.; Nazar, M.; Arbab, A.A.I.; Xu, T.; Yousif, M.H.; Mao, Y.; Yang, Z. Genome-Wide Association Study Identifies Candidate Genes Associated with Feet and Leg Conformation Traits in Chinese Holstein Cattle. Animals 2021, 11, 2259. [Google Scholar] [CrossRef] [PubMed]
- Galliou, J.M.; Kiser, J.N.; Oliver, K.F.; Seabury, C.M.; Moraes, J.G.N.; Burns, G.W.; Spencer, T.E.; Dalton, J.; Neibergs, H.L. Identification of Loci and Pathways Associated with Heifer Conception Rate in U.S. Holsteins. Genes 2020, 11, 767. [Google Scholar] [CrossRef]
- Atlasy, N.; Bujko, A.; Bækkevold, E.S.; Brazda, P.; Janssen-Megens, E.; Lundin, K.E.A.; Jahnsen, J.; Jahnsen, F.L.; Stunnenberg, H.G. Single Cell Transcriptomic Analysis of the Immune Cell Compartment in the Human Small Intestine and in Celiac Disease. Nat. Commun. 2022, 13, 4920. [Google Scholar] [CrossRef] [PubMed]
- Ogorevc, J.; Kunej, T.; Razpet, A.; Dovc, P. Database of Cattle Candidate Genes and Genetic Markers for Milk Production and Mastitis. Anim. Genet. 2009, 40, 832–851. [Google Scholar] [CrossRef]
- Sugimoto, M.; Fujikawa, A.; Womack, J.E.; Sugimoto, Y. Evidence That Bovine Forebrain Embryonic Zinc Finger-like Gene Influences Immune Response Associated with Mastitis Resistance. Proc. Natl. Acad. Sci. USA 2006, 103, 6454–6459. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, R.; Lu, Y.; Zou, X.; Yang, W. Aire and Fezf2, Two Regulators in Medullary Thymic Epithelial Cells, Control Autoimmune Diseases by Regulating TSAs: Partner or Complementer? Front. Immunol. 2022, 13, 948259. [Google Scholar] [CrossRef]
- Baltz, A.G.; Munschauer, M.; Schwanhäusser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The MRNA-Bound Proteome and Its Global Occupancy Profile on Protein-Coding Transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative A First Update on Mapping the Human Genetic Architecture of COVID-19. Nature 2022, 608, E1–E10. [CrossRef]
- Chen, Z.; Yao, Y.; Ma, P.; Wang, Q.; Pan, Y. Haplotype-Based Genome-Wide Association Study Identifies Loci and Candidate Genes for Milk Yield in Holsteins. PLoS ONE 2018, 13, e0192695. [Google Scholar] [CrossRef]
- Marete, A.; Lund, M.S.; Boichard, D.; Ramayo-Caldas, Y. A System-Based Analysis of the Genetic Determinism of Udder Conformation and Health Phenotypes across Three French Dairy Cattle Breeds. PLoS ONE 2018, 13, e0199931. [Google Scholar] [CrossRef] [PubMed]
- Tiezzi, F.; Maisano, A.M.; Chessa, S.; Luini, M.; Biffani, S. Heritability of Teat Condition in Italian Holstein Friesian and Its Relationship with Milk Production and Somatic Cell Score. Animals 2020, 10, 2271. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gudra, D.; Valdovska, A.; Jonkus, D.; Galina, D.; Kairisa, D.; Ustinova, M.; Viksne, K.; Fridmanis, D.; Kalnina, I. Genomic Characterization and Initial Insight into Mastitis-Associated SNP Profiles of Local Latvian Bos taurus Breeds. Animals 2023, 13, 2776. https://doi.org/10.3390/ani13172776
Gudra D, Valdovska A, Jonkus D, Galina D, Kairisa D, Ustinova M, Viksne K, Fridmanis D, Kalnina I. Genomic Characterization and Initial Insight into Mastitis-Associated SNP Profiles of Local Latvian Bos taurus Breeds. Animals. 2023; 13(17):2776. https://doi.org/10.3390/ani13172776
Chicago/Turabian StyleGudra, Dita, Anda Valdovska, Daina Jonkus, Daiga Galina, Daina Kairisa, Maija Ustinova, Kristine Viksne, Davids Fridmanis, and Ineta Kalnina. 2023. "Genomic Characterization and Initial Insight into Mastitis-Associated SNP Profiles of Local Latvian Bos taurus Breeds" Animals 13, no. 17: 2776. https://doi.org/10.3390/ani13172776
APA StyleGudra, D., Valdovska, A., Jonkus, D., Galina, D., Kairisa, D., Ustinova, M., Viksne, K., Fridmanis, D., & Kalnina, I. (2023). Genomic Characterization and Initial Insight into Mastitis-Associated SNP Profiles of Local Latvian Bos taurus Breeds. Animals, 13(17), 2776. https://doi.org/10.3390/ani13172776