
Genome Variation Map of Domestic Qinghai-Tibet Plateau Yaks by SLAF-Seq Reveals Genetic Footprint during Artificial Selection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Ethical Approval
2.2. Sample Collections, Sequencing, and SNP Calling
2.3. Phylogenetic Tree and Population Genetic Analyses
2.4. Fst Estimation
2.5. Protein Functional Annotation and GO Enrichment
2.6. π Ratio Estimates
2.7. Identifying Selective Sweep Regions
2.8. Gene Flow and Divergence Time Analysis
3. Results
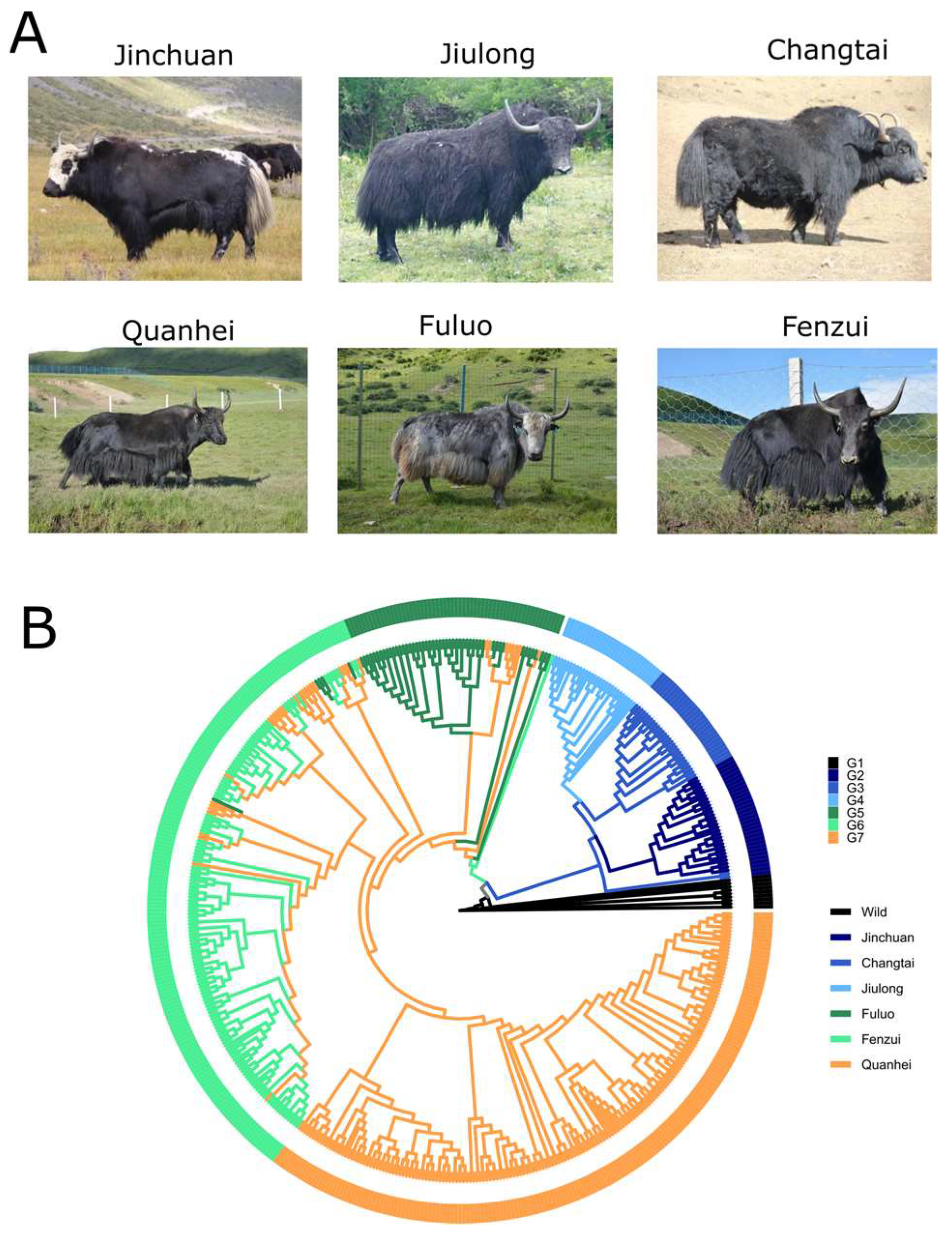
3.1. Genome Variation Map of Yaks
3.2. Phylogenetic Tree
3.3. Population Structure and Genetic Diversity
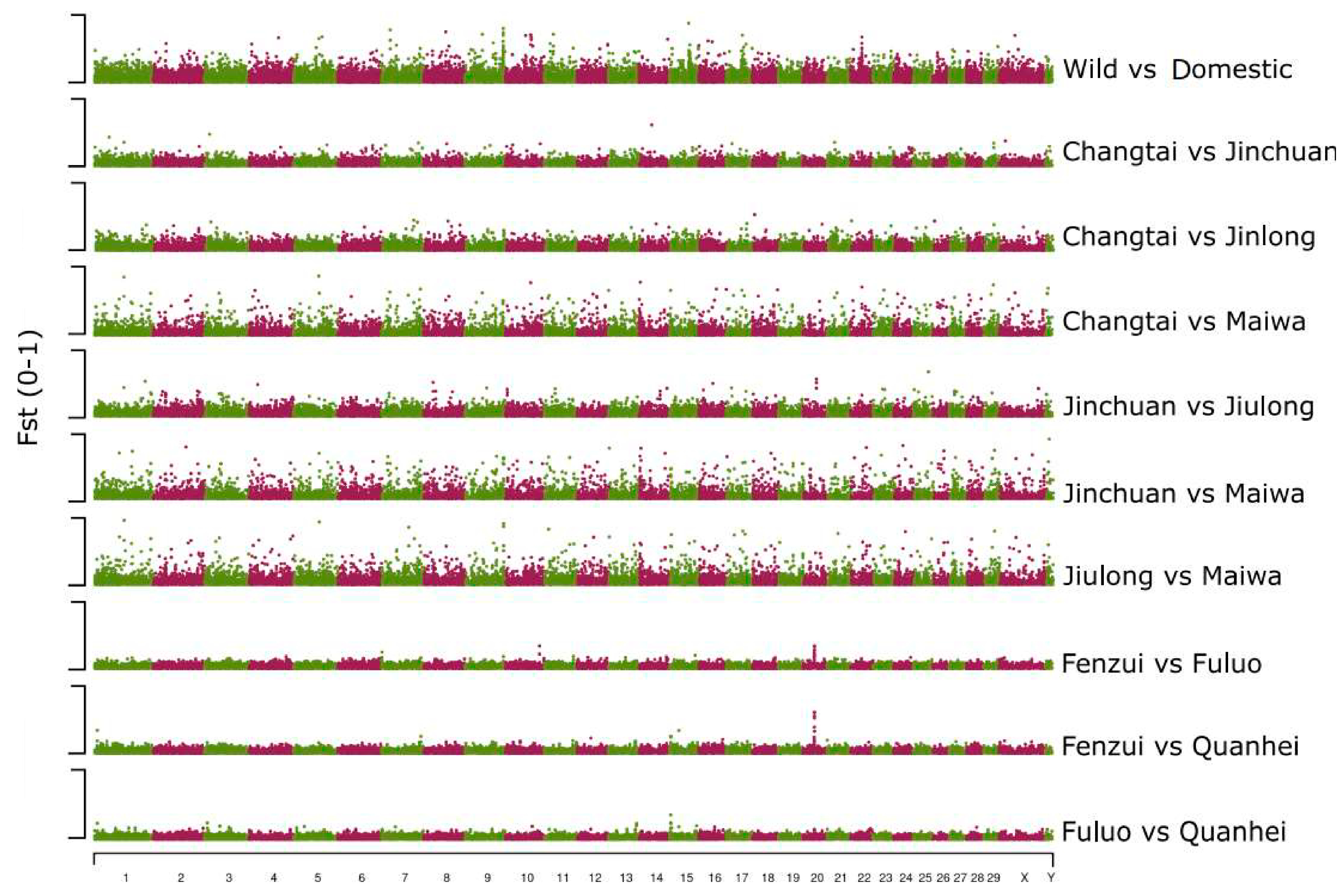
3.4. Genome-Wide Selection and Genomic Differentiation Analysis
3.5. Gene Flow and Divergence Time Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, K.; Lenstra, J.A.; Zhang, S.; Liu, W.; Liu, J. Evolution and domestication of the Bovini species. Anim. Genet. 2020, 51, 637–657. [Google Scholar] [CrossRef]
- Miao, F.; Guo, Z.; Xue, R.; Wang, X.; Shen, Y. Effects of grazing and precipitation on herbage biomass, herbage nutritive value, and yak performance in an alpine meadow on the Qinghai-Tibetan Plateau. PLoS ONE 2015, 10, e0127275. [Google Scholar] [CrossRef]
- Qiu, Q.; Wang, L.; Wang, K.; Yang, Y.; Ma, T.; Wang, Z.; Zhang, X.; Ni, Z.; Hou, F.; Long, R.; et al. Yak whole-genome resequencing reveals domestication signatures and prehistoric population expansions. Nat. Commun. 2015, 6, 10283. [Google Scholar] [CrossRef]
- Guo, S.; Savolainen, P.; Su, J.; Zhang, Q.; Qi, D.; Zhou, J.; Zhong, Y.; Zhao, X.; Liu, J. Origin of mitochondrial DNA diversity of domestic yaks. BMC Evol. Biol. 2006, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xin, S.; Liu, B.; Su, J.; Liu, J. Phylogeographical analyses of domestic and wild yaks based on mitochondrial DNA: New data and reappraisal. J. Biogeogr. 2010, 37, 2332–2344. [Google Scholar] [CrossRef]
- Jia, C.; Wang, H.; Li, C.; Wu, X.; Zan, L.; Ding, X.; Guo, X.; Bao, P.; Pei, J.; Chu, M.; et al. Genome-wide detection of copy number variations in polled yak using the Illumina BovineHD BeadChip. BMC Genom. 2019, 20, 376. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Hu, Q.; Ma, H.; Wang, L.; Yang, Y.; Luo, W.; Qiu, Q. Genome-wide variation within and between wild and domestic yak. Mol. Ecol. Resour. 2014, 14, 794–801. [Google Scholar] [CrossRef]
- Ellegren, H.; Sheldon, B.C. Genetic basis of fitness differences in natural populations. Nature 2008, 452, 169–175. [Google Scholar] [CrossRef]
- Stapley, J.; Reger, J.; Feulner, P.G.; Smadja, C.; Galindo, J.; Ekblom, R.; Bennison, C.; Ball, A.D.; Beckerman, A.P.; Slate, J. Adaptation genomics: The next generation. Trends Ecol. Evol. 2010, 25, 705–712. [Google Scholar] [CrossRef]
- Helyar, S.J.; Hemmer-Hansen, J.; Bekkevold, D.; Taylor, M.I.; Ogden, R.; Limborg, M.T.; Cariani, A.; Maes, G.E.; Diopere, E.; Carvalho, G.R.; et al. Application of SNPs for population genetics of nonmodel organisms: New opportunities and challenges. Mol. Ecol. Resour. 2011, 11 (Suppl. S1), 123–136. [Google Scholar] [CrossRef]
- Kerstens, H.H.; Crooijmans, R.P.; Veenendaal, A.; Dibbits, B.W.; Chin, A.W.T.F.; den Dunnen, J.T.; Groenen, M.A. Large scale single nucleotide polymorphism discovery in unsequenced genomes using second generation high throughput sequencing technology: Applied to turkey. BMC Genom. 2009, 10, 479. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Li, R.; Xu, X.; Jin, W.; Xu, M.; Zhao, H.; Xiang, Z.; Song, W.; Ying, K.; Zhang, M.; et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 2010, 42, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Larkin, D.M.; Daetwyler, H.D.; Hernandez, A.G.; Wright, C.L.; Hetrick, L.A.; Boucek, L.; Bachman, S.L.; Band, M.R.; Akraiko, T.V.; Cohen-Zinder, M.; et al. Whole-genome resequencing of two elite sires for the detection of haplotypes under selection in dairy cattle. Proc. Natl. Acad. Sci. USA 2012, 109, 7693–7698. [Google Scholar] [CrossRef]
- Lai, S.J.; Chen, S.Y.; Liu, Y.P.; Yao, Y.G. Mitochondrial DNA sequence diversity and origin of Chinese domestic yak. Anim. Genet. 2007, 38, 77–80. [Google Scholar] [CrossRef]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yonezawa, T.; Liu, B.; Ma, T.; Shen, X.; Su, J.; Guo, S.; Hasegawa, M.; Liu, J. Domestication relaxed selective constraints on the yak mitochondrial genome. Mol. Biol. Evol. 2011, 28, 1553–1556. [Google Scholar] [CrossRef]
- Chai, Z.X.; Xin, J.W.; Zhang, C.F.; Dawayangla; Luosang; Zhang, Q.; Pingcuozhandui; Li, C.; Zhu, Y.; Cao, H.W.; et al. Whole-genome resequencing provides insights into the evolution and divergence of the native domestic yaks of the Qinghai-Tibet Plateau. BMC Evol. Biol. 2020, 20, 137. [Google Scholar] [CrossRef]
- Ji, Q.M.; Xin, J.W.; Chai, Z.X.; Zhang, C.F.; Dawa, Y.; Luo, S.; Zhang, Q.; Pingcuo, Z.; Peng, M.S.; Zhu, Y.; et al. A chromosome-scale reference genome and genome-wide genetic variations elucidate adaptation in yak. Mol. Ecol. Resour. 2021, 21, 201–211. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, W.; Liu, X.; Du, X.; Zhang, K.; Zhang, Y.; Song, Y.; Zi, Y.; Qiu, Q.; Lenstra, J.A.; et al. Structural Variants Selected during Yak Domestication Inferred from Long-Read Whole-Genome Sequencing. Mol. Biol. Evol. 2021, 38, 3676–3680. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P. Variant Annotation and Functional Prediction: SnpEff. Methods Mol. Biol. 2022, 2493, 289–314. [Google Scholar] [CrossRef] [PubMed]
- Slifer, S.H. PLINK: Key Functions for Data Analysis. Curr. Protoc. Hum. Genet. 2018, 97, e59. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. Genome-wide complex trait analysis (GCTA): Methods, data analyses, and interpretations. Methods Mol. Biol. 2013, 1019, 215–236. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Hernández-Plaza, A.; Szklarczyk, D.; Botas, J.; Cantalapiedra, C.P.; Giner-Lamia, J.; Mende, D.R.; Kirsch, R.; Rattei, T.; Letunic, I.; Jensen, L.J.; et al. eggNOG 6.0: Enabling comparative genomics across 12535 organisms. Nucleic Acids Res. 2023, 51, D389–D394. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef]
- Schmutz, S.M. Genetics of coat color in cattle. In Bovine Genomics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Li, G.; Luo, J.; Wang, F.; Xu, D.; Ahmed, Z.; Chen, S.; Li, R.; Ma, Z. Whole-genome resequencing reveals genetic diversity, differentiation, and selection signatures of yak breeds/populations in Qinghai, China. Front. Genet. 2022, 13, 1034094. [Google Scholar] [CrossRef]
- Xia, X.; Yao, Y.; Li, C.; Zhang, F.; Qu, K.; Chen, H.; Huang, B.; Lei, C. Genetic diversity of Chinese cattle revealed by Y-SNP and Y-STR markers. Anim. Genet. 2019, 50, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Sabo, S.L.; Lahr, J.M.; Offer, M.; Weekes, A.; Sceniak, M.P. GRIN2B-related neurodevelopmental disorder: Current understanding of pathophysiological mechanisms. Front. Synaptic Neurosci. 2022, 14, 1090865. [Google Scholar] [CrossRef]
- Yang, R.; Walder-Christensen, K.K.; Kim, N.; Wu, D.; Lorenzo, D.N.; Badea, A.; Jiang, Y.H.; Yin, H.H.; Wetsel, W.C.; Bennett, V. ANK2 autism mutation targeting giant ankyrin-B promotes axon branching and ectopic connectivity. Proc. Natl. Acad. Sci. USA 2019, 116, 15262–15271. [Google Scholar] [CrossRef]
- Carbonell, A.U.; Cho, C.H.; Tindi, J.O.; Counts, P.A.; Jordan, B.A. Haploinsufficiency in the ANKS1B gene encoding AIDA-1 leads to a neurodevelopmental syndrome. Nat. Commun. 2019, 10, 3529. [Google Scholar] [CrossRef] [PubMed]
- Parcerisas, A.; Ortega-Gascó, A.; Pujadas, L.; Soriano, E. The Hidden Side of NCAM Family: NCAM2, a Key Cytoskeleton Organization Molecule Regulating Multiple Neural Functions. Int. J. Mol. Sci. 2021, 22, 10021. [Google Scholar] [CrossRef]
- Jensen, B.M.; Akin, C.; Gilfillan, A.M. Pharmacological targeting of the KIT growth factor receptor: A therapeutic consideration for mast cell disorders. Br. J. Pharmacol. 2008, 154, 1572–1582. [Google Scholar] [CrossRef]
- Lan, D.; Xiong, X.; Mipam, T.D.; Fu, C.; Li, Q.; Ai, Y.; Hou, D.; Chai, Z.; Zhong, J.; Li, J. Genetic Diversity, Molecular Phylogeny, and Selection Evidence of Jinchuan Yak Revealed by Whole-Genome Resequencing. G3-Genes Genom Genet. 2018, 8, 945–952. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Yang, J.; Liu, Y.; Jiang, M. Genome Variation Map of Domestic Qinghai-Tibet Plateau Yaks by SLAF-Seq Reveals Genetic Footprint during Artificial Selection. Animals 2023, 13, 2963. https://doi.org/10.3390/ani13182963
Li B, Yang J, Liu Y, Jiang M. Genome Variation Map of Domestic Qinghai-Tibet Plateau Yaks by SLAF-Seq Reveals Genetic Footprint during Artificial Selection. Animals. 2023; 13(18):2963. https://doi.org/10.3390/ani13182963
Chicago/Turabian StyleLi, Biao, Jinzeng Yang, Yili Liu, and Mingfeng Jiang. 2023. "Genome Variation Map of Domestic Qinghai-Tibet Plateau Yaks by SLAF-Seq Reveals Genetic Footprint during Artificial Selection" Animals 13, no. 18: 2963. https://doi.org/10.3390/ani13182963
APA StyleLi, B., Yang, J., Liu, Y., & Jiang, M. (2023). Genome Variation Map of Domestic Qinghai-Tibet Plateau Yaks by SLAF-Seq Reveals Genetic Footprint during Artificial Selection. Animals, 13(18), 2963. https://doi.org/10.3390/ani13182963