
Variations in the Intestinal Microbiota of the Chinese Soft-Shelled Turtle (Trionyx sinensis) between Greenhouse and Pond Aquaculture
 , , , and
, , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. DNA Extraction, PCR Amplification, and MiSeq Sequencing
2.3. 16S rRNA Gene Analysis
2.4. Statistical Analysis
3. Results
3.1. Characteristics of 16S rRNA Gene Sequencing
3.2. Differences in the Bacterial Community Compositions
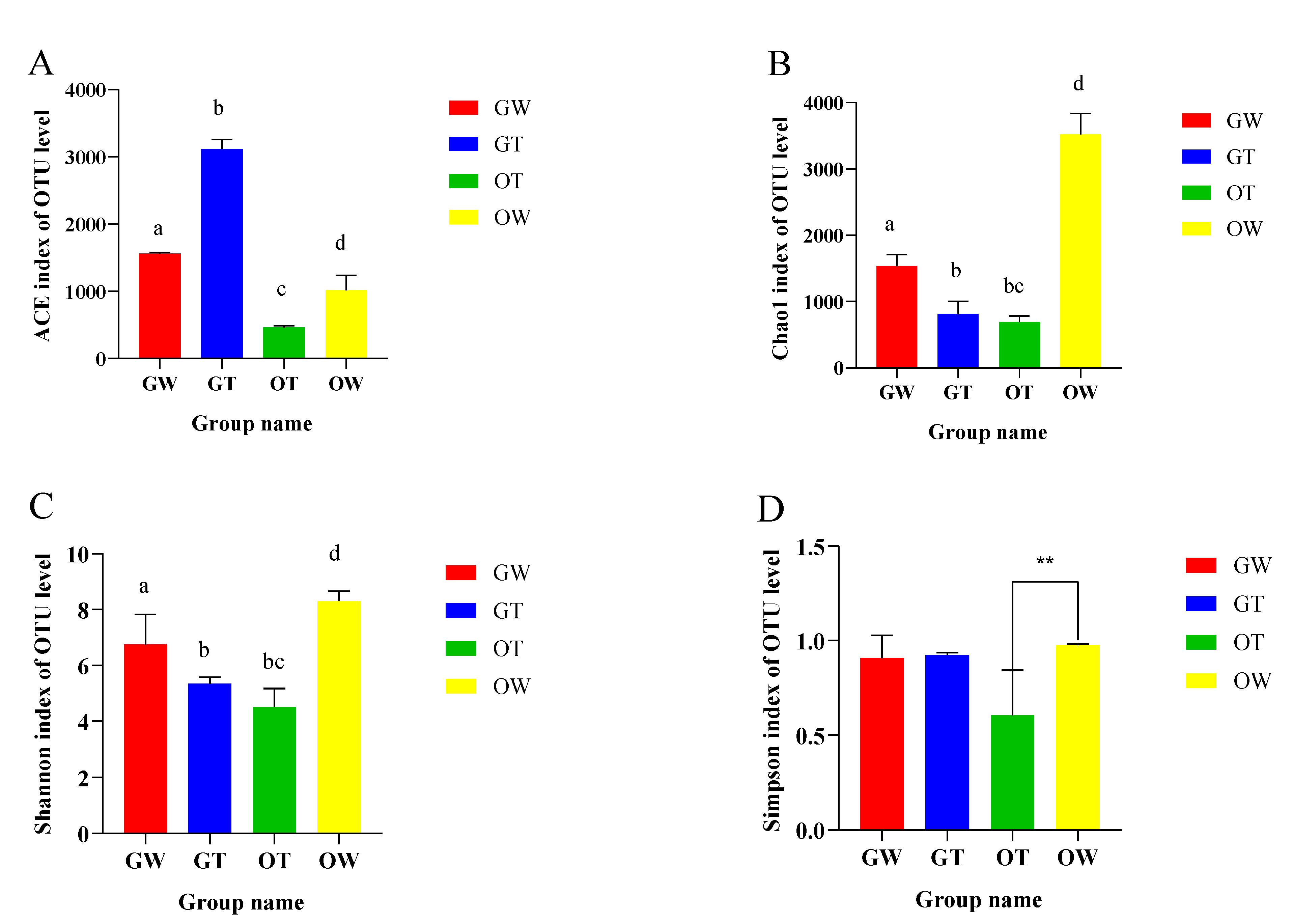
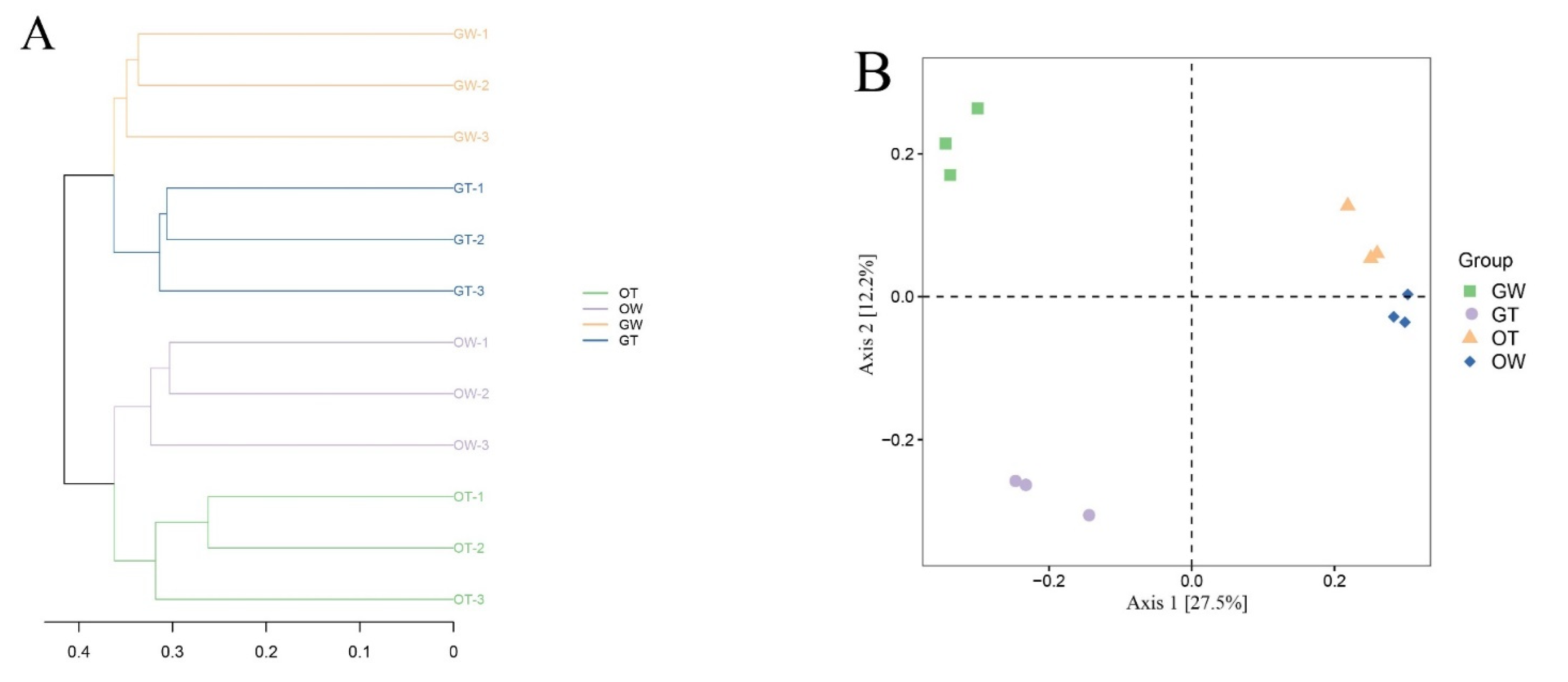
3.2.1. Differences in Intestinal Microbiota between Greenhouse and Pond Aquacultures of SSTs
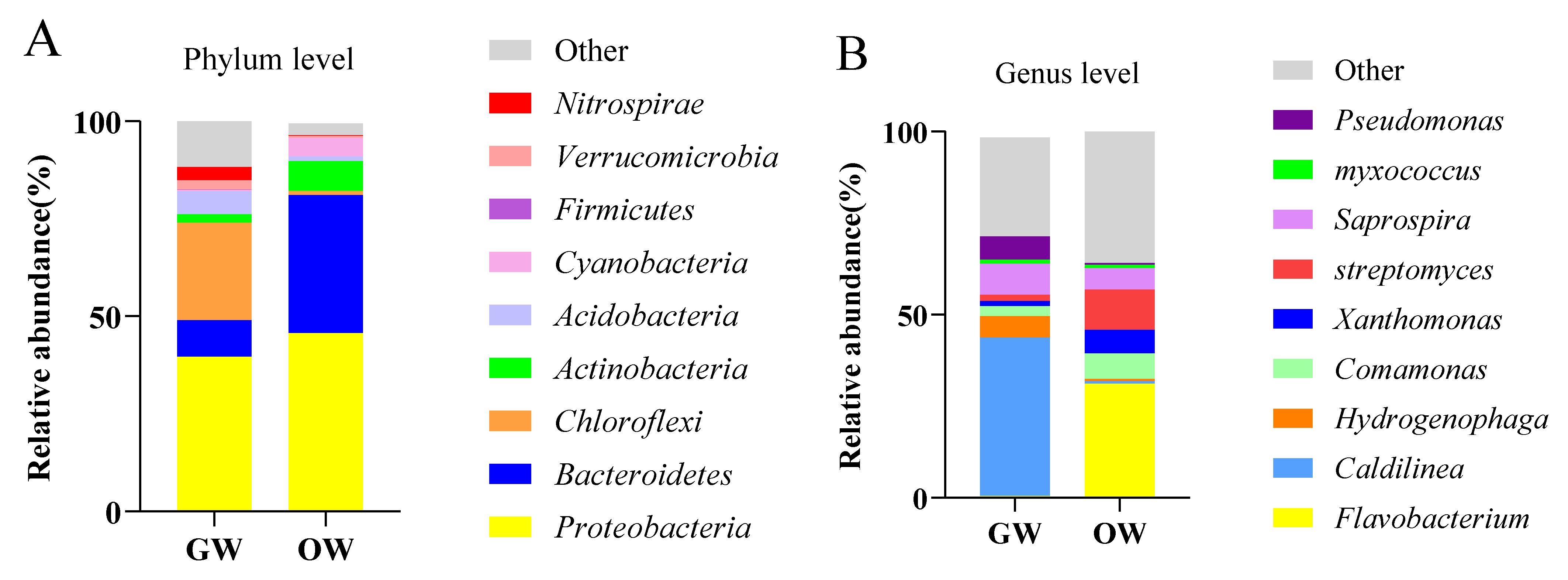
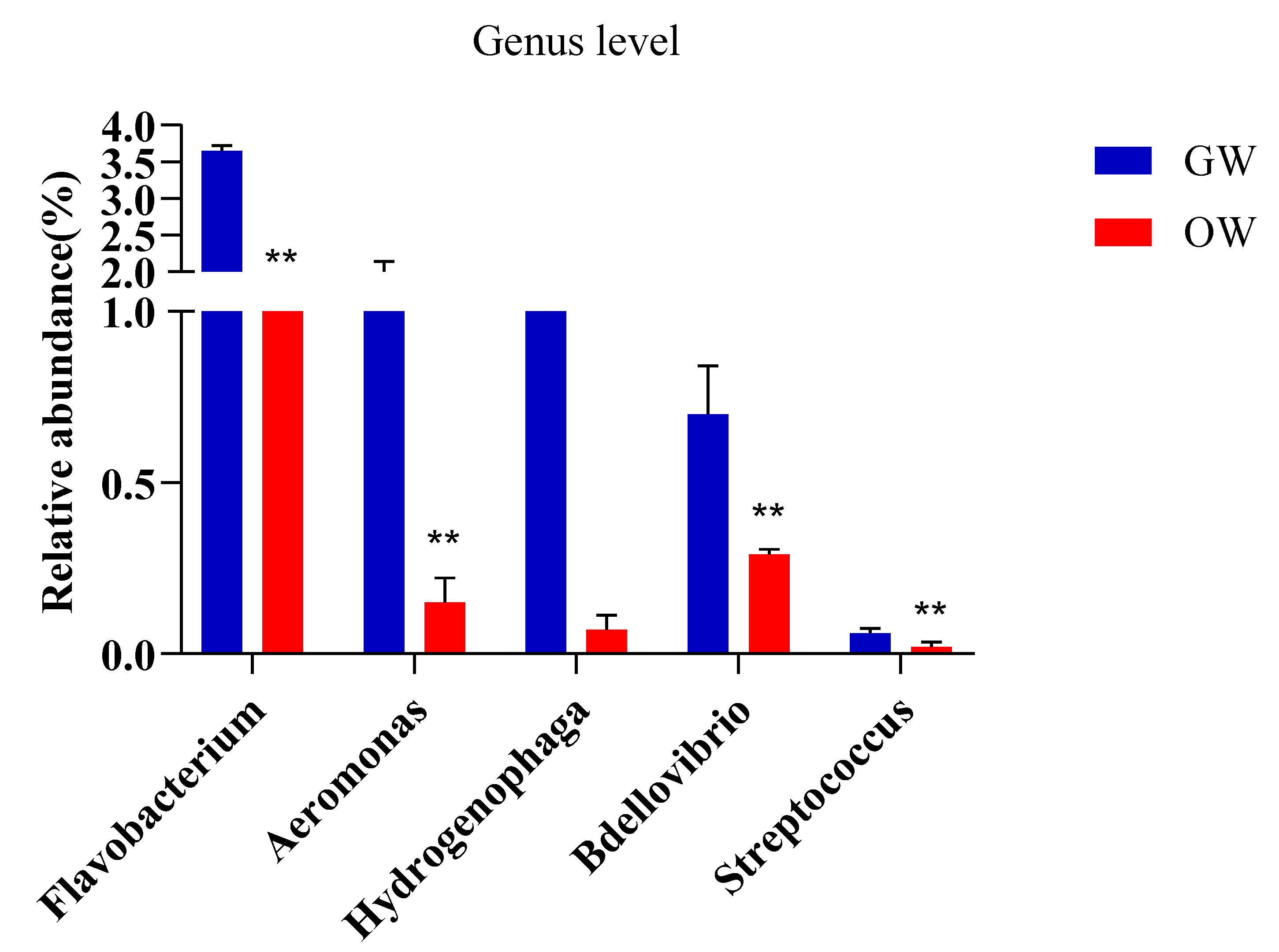
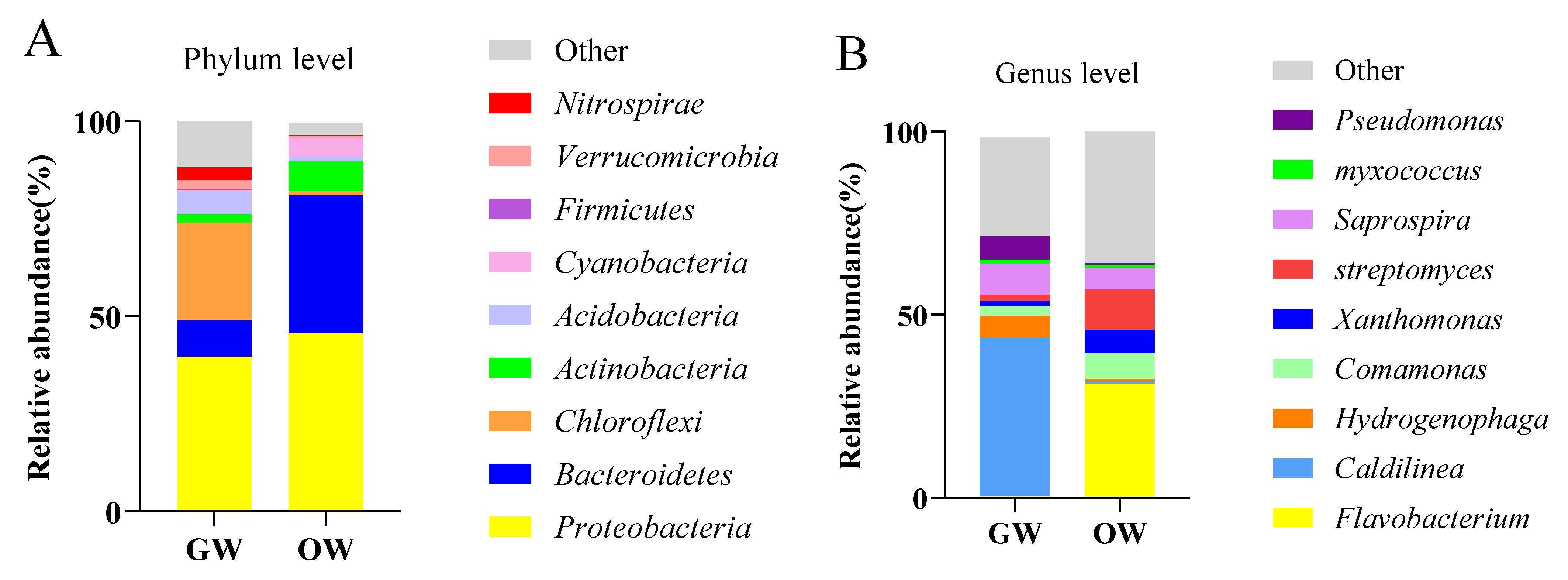
3.2.2. Differences in Bacterial Community Composition between Greenhouse and Pond Cultures of SSTs
3.3. Environmental Impact on the Changes of Intestinal Flora in SSTs
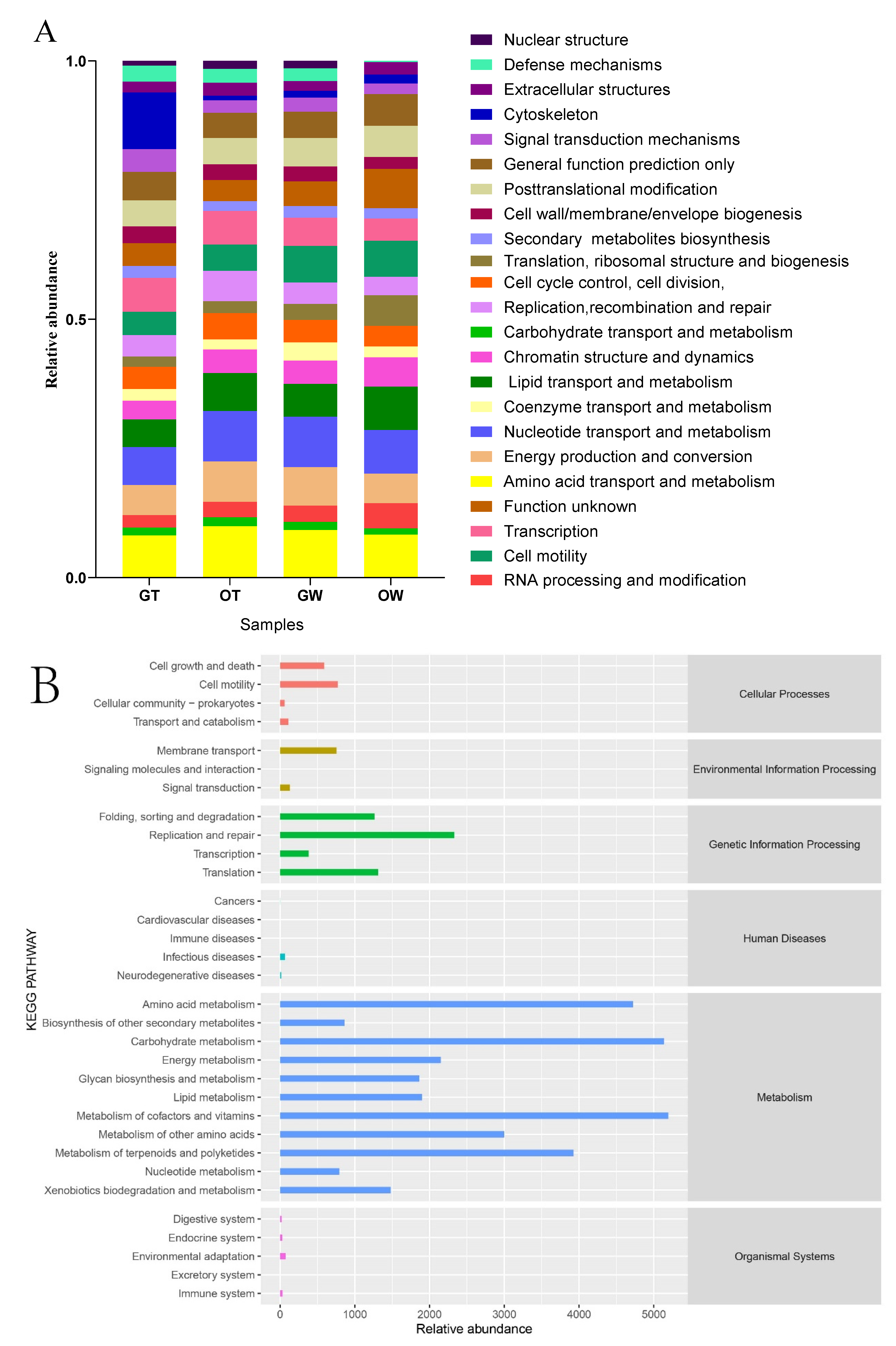
3.4. Functional Prediction of the Microbiota
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, T.F.; Han, Y.W.; Chen, S.L.; Zhao, H.Y. Genome-wide identification of Toll like receptors in the Chinese soft-shelled turtle (Pelodiscus sinensis) and expression analysis responding to Aeromonas hydrophila infection. Fish Shellfish Immunol. 2019, 87, 478–489. [Google Scholar] [CrossRef]
- Wu, Y.C.; Liu, X.; Wang, J.L.; Chen, X.L.; Lei, L.; Han, J.; Jiang, Y.S.; Ling, Z.Q. Soft-shelled turtle peptide modulates microRNA profile in human gastric cancer AGS cells. Oncol. Lett. 2018, 15, 3109–3120. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jiang, G.M.; Peng, Z.T.; Li, Y.L.; Li, J.L.; Zou, L.; He, Z.G.; Wang, X.Q.; Chu, W.Y. The effect of high fat diet on daily rhythm of the core clock genes and muscle functional genes in the skeletal muscle of Chinese soft-shelled turtle (Trionyx sinensis). Comp. Biochem. Physiol. B-Biochem. Mol. Biol. 2017, 213, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lyu, S.J.; Yuan, X.M.; Zhang, H.Q.; Hang, X.Y.; Li, Y.L.; Shi, W.D.; Liu, L.; Yu, Z.; Wu, Y.L. Transcriptome profiling analysis of lung tissue of Chinese soft-shell turtle infected by Trionyx sinensis Hemorrhagic Syndrome Virus. Fish Shellfish Immunol. 2020, 98, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Guo, Q.L.; Dai, H.P. Identification of differentially expressed immune-relevant genes in Chinese soft-shelled turtle (Trionyx sinensis) infected with Aeromonas hydrophila. Vet. Immunol. Immunopathol. 2008, 125, 82–91. [Google Scholar] [CrossRef]
- Tokita, M.; Kuratani, S. Normal embryonic stages of the Chinese softshelled turtle (Pelodiscus sinensis). Zool. Sci. 2001, 18, 705–715. [Google Scholar] [CrossRef]
- Defoirdt, T.; Sorgeloos, P.; Bossier, P. Alternatives to antibiotics for the control of bacterial disease in aquaculture. Curr. Opin. Microbiol. 2011, 14, 251–258. [Google Scholar] [CrossRef]
- Blancheton, J.P.; Attramadal, K.J.K.; Michaud, L.; d’Orbcastel, E.R.; Vadstein, O. Insight into bacterial population in aquaculture systems and its implication. Aquac. Eng. 2013, 53, 30–39. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, W.; Xiao, L.; Yang, L.Y. Effect of decabromodiphenyl ether (BDE 209) and dibromodiphenyl ether (BDE 15) on soil microbial activity and bacterial community composition. J. Hazard. Mater. 2011, 186, 883–890. [Google Scholar] [CrossRef]
- Xiong, J.B.; Nie, L.; Chen, J. Current understanding on the roles of intestinal microbiota in fish disease and immunity. Zool. Res. 2019, 40, 70–76. [Google Scholar] [PubMed]
- Eisenstein, M. The hunt for a healthy microbiome. Nature 2020, 577, S6–S8. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.F.; Wang, Z.L.; Chen, M.S.; Qu, Y.X.; Li, J.Y.; Zhou, A.G.; Xie, S.L.; Zeng, F.; Zou, J.X. Microbiota comparison of Pacific white shrimp intestine and sediment at freshwater and marine cultured environment. Sci. Total Environ. 2019, 657, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Yokozawa, J.; Tsuda, K.; Ito, P.X.Q.; Nagasaki, M.; Yasuda, J. An efficient quantitation method of next-generation sequencing libraries by using MiSeq sequencer. Anal. Biochem. 2014, 466, 27–29. [Google Scholar] [CrossRef]
- Xue, M.; Wu, Y.; Hong, Y.; Meng, Y.; Xu, C.; Jiang, N.; Li, Y.; Liu, W.; Fan, Y.; Zhou, Y. Effects of dietary Bacillus amyloliquefaciens on the growth, immune responses, intestinal microbiota composition and disease resistance of yellow catfish, (Pelteobagrus fulvidraco). Front. Cell Infect. Microbiol. 2022, 12, 7351. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Gaskins, H.R.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- Moutselos, K.; Kanaris, I.; Chatziioannou, A.; Maglogiannis, I.; Kolisis, F.N. KEGGconverter: A tool for the in-silico modelling of metabolic networks of the KEGG Pathways database. BMC Bioinform. 2009, 10, 1123156. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the Effects of PCR Amplification and Sequencing Artifacts on 16S rRNA-Based Studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef]
- Abraham, T.J.; Ghosh, S.; Nagesh, T.S.; Sasmal, D. Distribution of bacteria involved in nitrogen and sulphur cycles in shrimp culture systems of West Bengal, India. Aquaculture 2004, 239, 275–288. [Google Scholar] [CrossRef]
- Cuellar-Gempeler, C.; Leibold, M.A. Multiple colonist pools shape fiddler crab-associated bacterial communities. ISME J. 2018, 12, 825–837. [Google Scholar] [CrossRef]
- Giatsis, C.; Sipkema, D.; Smidt, H.; Verreth, J.; Verdegem, M. The Colonization Dynamics of the Intestinal Microbiota in Tilapia Larvae. PLoS ONE 2014, 9, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 432342. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.E.; Field, D.; Gilbert, J.A. Predicting bacterial community assemblages using an artificial neural network approach. Nat. Methods 2012, 9, 621. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.J.; Cronan, J.E. Temperature regulation of membrane composition in the Firmicute, Enterococcus faecalis, parallels that of Escherichia coli. Environ. Microbiol. 2021, 23, 2683–2691. [Google Scholar] [CrossRef]
- Li, S.; Chen, T.; Dong, S.; Xiong, Y.; Wei, H.; Xu, F. The Effects of Rebaudioside A on Microbial Diversity in Mouse Intestine. Food Sci. Technol. Res. 2014, 20, 459–467. [Google Scholar] [CrossRef]
- Shu, D.T.; He, Y.L.; Yue, H.; Wang, Q.Y. Microbial structures and community functions of anaerobic sludge in six full-scale wastewater treatment plants as revealed by 454 high-throughput pyrosequencing. Bioresour. Technol. 2015, 186, 163–172. [Google Scholar] [CrossRef]
- Klase, G.; Lee, S.; Liang, S.; Kim, J.; Zo, Y.G.; Lee, J. The microbiome and antibiotic resistance in integrated fishfarm water: Implications of environmental public health. Sci. Total Environ. 2019, 649, 1491–1501. [Google Scholar] [CrossRef]
- Zhang, J.X.; Yang, Y.Y.; Zhao, L.; Li, Y.Z.; Xie, S.G.; Liu, Y. Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Appl. Microbiol. Biotechnol. 2015, 99, 3291–3302. [Google Scholar] [CrossRef]
- Gao, S.; Pan, L.Q.; Huang, F.; Song, M.S.; Tian, C.C.; Zhang, M.Y. Metagenomic insights into the structure and function of intestinal microbiota of the farmed Pacific white shrimp (Litopenaeus vannamei). Aquaculture 2019, 499, 109–118. [Google Scholar] [CrossRef]
- Li, E.C.; Xu, C.; Wang, X.D.; Wang, S.F.; Zhao, Q.; Zhang, M.L.; Qin, J.G.; Chen, L.Q. Intestinal Microbiota and its Modulation for Healthy Farming of Pacific White Shrimp Litopenaeus vannamei. Fish. Sci. Aquac. 2018, 26, 381–399. [Google Scholar]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the intestinal microbiota: Relationship to diet, obesity and time in mouse models. Intestinal 2010, 59, 1635–1642. [Google Scholar]
- Shang, Y.; Kumar, S.; Oakley, B.; Kim, W.K. Chicken Intestinal Microbiota: Importance and Detection Technology. Front. Vet. Sci. 2018, 5, 1232. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.; Singh, G.; O-Sullivan, I.; Ma, K.G.; Anbazhagan, A.N.; Votta-Velis, E.G.; Bruce, B.; Richard, R.; van Wijnen, A.J.; Im, H.J. Intestinal-microbiota modulation: The impact of the intestinal-microbiota on osteoarthritis. Gene 2021, 785, 143213. [Google Scholar] [CrossRef] [PubMed]
- Van Keulen, G.; Dyson, P.J. Production of Specialized Metabolites by Streptomyces coelicolor A3. In Advances in Applied Microbiology; Sariaslani, S., Gadd, G.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 89, pp. 217–266. [Google Scholar]
- Binda, C.; Lopetuso, L.R.; Rizzatti, G.; Gibiino, G.; Cennamo, V.; Gasbarrini, A. Actinobacteria: A relevant minority for the maintenance of intestinal homeostasis. Dig. Liver Dis. 2018, 50, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.X.; Wu, S.S.; Zeng, Z.Y.; Fu, Z.W. Effects of environmental pollutants on intestinal microbiota. Environ. Pollut. 2017, 222, 1–9. [Google Scholar] [CrossRef]
- Qiu, Y.J.; Kidera, N.; Hayashi, M.; Fujishima, K.; Tamura, H. Rickettsia spp. and Ehrlichia spp. in Amblyomma ticks parasitizing wild amphibious sea kraits and yellow-margined box turtles in Okinawa, Japan. Ticks Tick-Borne Dis. 2021, 12, 221–229. [Google Scholar] [CrossRef]
- Adams, D.M.; Williamson, S.A.; Evans, R.G.; Reina, R.D. Increasing hypoxia progressively slows early embryonic development in an oviparous reptile, the green turtle, Chelonia mydas. R. Soc. Open Sci. 2022, 9, 8. [Google Scholar] [CrossRef]
- Campos, P.; Guivernau, M.; Prenafeta-Boldu, F.X.; Cardona, L. Fast acquisition of a polysaccharide fermenting intestinal microbiome by juvenile green turtles Chelonia mydas after settlement in coastal habitats. Microbiome 2018, 6, 123112. [Google Scholar] [CrossRef]
- Stecher, B.; Maier, L.; Hardt, W.D. ‘Blooming’ in the intestinal: How dysbiosis might contribute to pathogen evolution. Nat. Rev. Microbiol. 2013, 11, 277–284. [Google Scholar] [CrossRef]
- Yan, Q.Y.; Li, J.J.; Yu, Y.H.; Wang, J.J.; He, Z.L.; Van Nostrand, J.D.; Kempher, M.L.; Wu, L.Y.; Wang, Y.P.; Liao, L.J.; et al. Environmental filtering decreases with fish development for the assembly of intestinal microbiota. Environ. Microbiol. 2016, 18, 4739–4754. [Google Scholar] [CrossRef]
- Baldo, L.; Riera, J.L.; Tooming-Klunderud, A.; Alba, M.M.; Salzburger, W. Intestinal Microbiota Dynamics during Dietary Shift in Eastern African Cichlid Fishes. PLoS ONE 2015, 10, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Han, X.Y.; Wei, D.; Xu, Z.R. Influence of Cu2+-loaded silicate on the growth performance and microflora of crucian carp Carassius auratus. Dis. Aquat. Org. 2009, 85, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.R.; Niu, C.J.; Pu, L.J. Effects of stocking density on growth and non-specific immune responses in juvenile soft-shelled turtle, (Pelodiscus sinensis). Aquac. Res. 2007, 38, 1380–1386. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on intestinal microbial diversity. Intestinal 2014, 63, 1913–1920. [Google Scholar]
- Serra, C.R.; Magalhaes, F.; Couto, A.; Oliva-Teles, A.; Enes, P. Intestinal microbiota and intestinal morphology of gilthead sea bream (Sparus aurata) juveniles are not affected by chromic oxide as digestibility marker. Aquac. Res. 2018, 49, 1347–1356. [Google Scholar] [CrossRef]
- Wu, B.L.; Luo, S.; Wang, J.W. Effects of temperature and feeding frequency on ingestion and growth for rare minnow. Physiol. Behav. 2015, 140, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Zhu, Q.J.; Xiao, F.R.; Ngwawa, J.M.; Zhang, H.X.; Shi, H.T. Preliminary Diet Analysis of Chinese Soft-shelled Turtle (Pelodiscus sinensis) in the Middle Confines of the Yellow River, Shaanxi Province, China. Pak. J. Zool. 2022, 54, 1489–1491. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, L.P.; Shen, Y.Q. A systematic review of advances in intestinal microflora of fish. Fish Physiol. Biochem. 2021, 47, 2041–2053. [Google Scholar] [CrossRef]
- Shang, P.; Dong, S.X.; Han, Y.Q.; Bo, S.X.; Ye, Y.R.; Duan, M.Q.; Chamba, Y. Environmental exposure to swine farms reshapes human intestinal microbiota. Chemosphere 2022, 307, 135558. [Google Scholar] [CrossRef]
- Sun, F.L.; Xu, Z.T. Significant Differences in Intestinal Microbial Communities in Aquatic Animals from an Aquaculture Area. J. Mar. Sci. Eng. 2021, 9, 104. [Google Scholar] [CrossRef]
- Tomasello, G.; Sorce, A.; Damiani, P.; Sinagra, E.; Carini, F. The importance of intestinal microbial flora (microbiota) and role of diet. Prog. Nutr. 2017, 19, 342–344. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Read Number | Coverage | Number of OTUs | Alpha Diversity | |||
|---|---|---|---|---|---|---|---|
| ACE | Chao1 | Shannon | Simpson | ||||
| GW-1 | 77,652 | 0.994 | 799 | 1574 | 1701.85 | 6.881 | 0.969 |
| GW-2 | 70,499 | 0.993 | 728 | 1574 | 1359.44 | 7.754 | 0.984 |
| GW-3 | 85,369 | 0.998 | 797 | 1551 | 1552.8 | 5.626 | 0.769 |
| GT-1 | 83,691 | 0.992 | 869 | 1239.7 | 875.423 | 5.338 | 0.933 |
| GT-2 | 85,337 | 0.994 | 887 | 1017.8 | 603.203 | 5.141 | 0.910 |
| GT-3 | 89,218 | 0.997 | 1039 | 798.3 | 963.684 | 5.586 | 0.929 |
| OW-1 | 97,192 | 0.991 | 649 | 3068.3 | 3879.18 | 8.572 | 0.984 |
| OW-2 | 96,507 | 0.997 | 730 | 3273.3 | 3403.96 | 8.429 | 0.969 |
| OW-3 | 96,507 | 0.993 | 621 | 3007.7 | 3272.43 | 7.911 | 0.972 |
| OW1-1 | 93,462 | 0.996 | 667 | 3223.2 | 3434.18 | 8.554 | 0.997 |
| OW1-2 | 96,757 | 0.994 | 754 | 3231.3 | 3334.76 | 8.664 | 0.965 |
| OW1-3 | 98,327 | 0.996 | 665 | 3643.4 | 3232.13 | 7.443 | 0.976 |
| OW2-1 | 95,462 | 0.994 | 665 | 3231.5 | 3443.38 | 8.334 | 0.955 |
| OW2-2 | 98,767 | 0.993 | 768 | 3431.3 | 3654.34 | 8.445 | 0.977 |
| OW2-3 | 91,347 | 0.993 | 643 | 3332.7 | 3121.12 | 7.944 | 0.966 |
| OT-1 | 82,997 | 0.997 | 884 | 491.4 | 796.035 | 3.888 | 0.456 |
| OT-2 | 86,878 | 0.995 | 789 | 465.1 | 664.27 | 4.423 | 0.475 |
| OT-3 | 74,136 | 0.998 | 932 | 437.7 | 617.694 | 5.219 | 0.880 |
| OT1-1 | 83,452 | 0.995 | 765 | 445.5 | 787.033 | 3.867 | 0.454 |
| OT1-2 | 83,423 | 0.996 | 865 | 422.3 | 654.45 | 4.455 | 0.487 |
| OT1-3 | 74,323 | 0.993 | 912 | 445.5 | 634.636 | 5.209 | 0.867 |
| OT2-1 | 84,432 | 0.994 | 877 | 488.6 | 756.086 | 3.888 | 0.476 |
| OT2-2 | 87,765 | 0.995 | 765 | 455.3 | 666.87 | 4.477 | 0.476 |
| OT2-3 | 75,546 | 0.997 | 911 | 444.7 | 636.677 | 5.256 | 0.855 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, N.; Zhang, P.; Xue, M.; Xiao, Z.; Zhang, M.; Meng, Y.; Fan, Y.; Qiu, J.; Zhang, Q.; Zhou, Y. Variations in the Intestinal Microbiota of the Chinese Soft-Shelled Turtle (Trionyx sinensis) between Greenhouse and Pond Aquaculture. Animals 2023, 13, 2971. https://doi.org/10.3390/ani13182971
Liu N, Zhang P, Xue M, Xiao Z, Zhang M, Meng Y, Fan Y, Qiu J, Zhang Q, Zhou Y. Variations in the Intestinal Microbiota of the Chinese Soft-Shelled Turtle (Trionyx sinensis) between Greenhouse and Pond Aquaculture. Animals. 2023; 13(18):2971. https://doi.org/10.3390/ani13182971
Chicago/Turabian StyleLiu, Naicheng, Peng Zhang, Mingyang Xue, Zidong Xiao, Mengjie Zhang, Yan Meng, Yuding Fan, Junqiang Qiu, Qinghua Zhang, and Yong Zhou. 2023. "Variations in the Intestinal Microbiota of the Chinese Soft-Shelled Turtle (Trionyx sinensis) between Greenhouse and Pond Aquaculture" Animals 13, no. 18: 2971. https://doi.org/10.3390/ani13182971
APA StyleLiu, N., Zhang, P., Xue, M., Xiao, Z., Zhang, M., Meng, Y., Fan, Y., Qiu, J., Zhang, Q., & Zhou, Y. (2023). Variations in the Intestinal Microbiota of the Chinese Soft-Shelled Turtle (Trionyx sinensis) between Greenhouse and Pond Aquaculture. Animals, 13(18), 2971. https://doi.org/10.3390/ani13182971