Adaptive Evolution of the OAS Gene Family Provides New Insights into the Antiviral Ability of Laurasiatherian Mammals


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Dataset Preparation and Sequence Alignments
2.2. Molecular Evolutionary Analyses
3. Results
3.1. OAS Gene Identification and Gene Tree Reconstruction
3.2. Selection Characteristics of OAS Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartmann, R.; Justesen, J.; Sarkar, S.N.; Sen, G.C.; Yee, V.C. Crystal structure of the 2’-specific and double-stranded RNA-activated interferon-induced antiviral protein 2’-5’-oligoadenylate synthetase. Mol. Cell 2003, 12, 1173–1185. [Google Scholar] [CrossRef]
- Zhao, L.; Jha, B.K.; Wu, A.; Elliott, R.; Ziebuhr, J.; Gorbalenya, A.E.; Silverman, R.H.; Weiss, S.R. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Amp. Microbe 2012, 11, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.; Schwerk, J.; Lim, C.; Kell, A.; Jarret, A.; Pangallo, J.; Loo, Y.-M.; Liu, S.; Hagedorn, C.H.; Gale, M.; et al. Interferon lambda 4 expression is suppressed by the host during viral infection. J. Exp. Med. 2016, 213, 2539–2552. [Google Scholar] [CrossRef] [Green Version]
- Trilling, M.; Bellora, N.; Rutkowski, A.J.; De Graaf, M.; Dickinson, P.; Robertson, K.; Prazeres da Costa, O.; Ghazal, P.; Friedel, C.C.; Albà, M.M.; et al. Deciphering the modulation of gene expression by type I and II interferons combining 4sU-tagging, translational arrest and in silico promoter analysis. Nucleic Acids Res. 2013, 41, 8107–8125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Jiang, X.; Pfau, D.; Ling, Y.; Nathan, C.F. Type I interferon signaling mediates Mycobacterium tuberculosis-induced macrophage death. J. Exp. Med. 2021, 218, e20200887. [Google Scholar] [CrossRef] [PubMed]
- Van Der Pouw Kraan, T.C.T.M.; Wijbrandts, C.A.; Van Baarsen, L.G.M.; Voskuyl, A.E.; Rustenburg, F.; Baggen, J.M.; Ibrahim, S.M.; Fero, M.; Dijkmans, B.A.C.; Tak, P.P.; et al. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: Assignment of a type I interferon signature in a subpopulation of patients. Ann. Rheum. Dis. 2007, 66, 1008–1014. [Google Scholar] [CrossRef]
- Kristiansen, H.; Gad, H.H.; Eskildsen-Larsen, S.; Despres, P.; Hartmann, R. The oligoadenylate synthetase family: An ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2011, 31, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H. Viral encounters with 2’,5’-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.K.; et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Mitnik, C.; Valente, G.; Floyd-Smith, G. Expansion and molecular evolution of the interferon-induced 2’-5’ oligoadenylate synthetase gene family. Mol. Biol. Evol. 2000, 17, 738–750. [Google Scholar] [CrossRef]
- Hancks, D.C.; Hartley, M.K.; Hagan, C.; Clark, N.L.; Elde, N.C. Overlapping Patterns of Rapid Evolution in the Nucleic Acid Sensors cGAS and OAS1 Suggest a Common Mechanism of Pathogen Antagonism and Escape. PLoS Genet. 2015, 11, e1005203. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Zhou, X.; Tian, L.; Wang, J.; Zhang, W. IFN-β1b induces OAS3 to inhibit EV71 via IFN-β1b/JAK/STAT1 pathway. Virol. Sin. 2022, 37, 676–684. [Google Scholar] [CrossRef]
- Mozzi, A.; Pontremoli, C.; Forni, D.; Clerici, M.; Pozzoli, U.; Bresolin, N.; Cagliani, R.; Sironi, M. OASes and STING: Adaptive evolution in concert. Genome Biol. Evol. 2015, 7, 1016–1032. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Boldrup, L.; Coates, P.J.; Fahraeus, R.; Nylander, E.; Loizou, C.; Olofsson, K.; Norberg-Spaak, L.; Gärskog, O.; Nylander, K. Epigenetic regulation of OAS2 shows disease-specific DNA methylation profiles at individual CpG sites. Sci. Rep. 2016, 6, 32579. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Hu, J.; Hu, Y.; Li, Y.; Xu, D.; Ryder, O.A.; Irwin, D.M.; Yu, L. Diverse phylogenomic datasets uncover a concordant scenario of laurasiatherian interordinal relationships. Mol. Phylogenet. Evol. 2021, 157, 107065. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.A.; Tachedjian, M.; Cui, J.; Cheng, A.Z.; Johnson, A.; Baker, M.L.; Harris, R.S.; Wang, L.-F.; Tachedjian, G. Differential Evolution of Antiretroviral Restriction Factors in Pteropid Bats as Revealed by APOBEC3 Gene Complexity. Mol. Biol. Evol. 2018, 35, 1626–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, K.; Morand, S.; Wardeh, M.; Baylis, M. Distinct spread of DNA and RNA viruses among mammals amid prominent role of domestic species. Glob. Ecol. Biogeogr. A J. Macroecol. 2020, 29, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Erratum: Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 548, 612. [Google Scholar] [CrossRef] [Green Version]
- Tollis, M.; Robbins, J.; Webb, A.E.; Kuderna, L.F.K.; Caulin, A.F.; Garcia, J.D.; Bèrubè, M.; Pourmand, N.; Marques-Bonet, T.; O’Connell, M.J.; et al. Return to the Sea, Get Huge, Beat Cancer: An Analysis of Cetacean Genomes Including an Assembly for the Humpback Whale (Megaptera novaeangliae). Mol. Biol. Evol. 2019, 36, 1746–1763. [Google Scholar] [CrossRef]
- Harvell, C.D.; Montecino-Latorre, D.; Caldwell, J.M.; Burt, J.M.; Bosley, K.; Keller, A.; Heron, S.F.; Salomon, A.K.; Lee, L.; Pontier, O.; et al. Disease epidemic and a marine heat wave are associated with the continental-scale collapse of a pivotal predator (Pycnopodia helianthoides). Sci. Adv. 2019, 5, eaau7042. [Google Scholar] [CrossRef]
- Wobessi, J.N.S.; Kenmoe, S.; Mahamat, G.; Belobo, J.T.E.; Emoh, C.P.D.; Efietngab, A.N.; Bebey, S.R.K.; Ngongang, D.T.; Tchatchouang, S.; Nzukui, N.D.; et al. Incidence and seroprevalence of rabies virus in humans, dogs and other animal species in Africa, a systematic review and meta-analysis. One Health 2021, 13, 100285. [Google Scholar] [CrossRef] [PubMed]
- Fooks, A.R.; Cliquet, F.; Finke, S.; Freuling, C.; Hemachudha, T.; Mani, R.S.; Müller, T.; Nadin-Davis, S.; Picard-Meyer, E.; Wilde, H.; et al. Rabies. Nat. Rev. Dis. Prim. 2017, 3, 17091. [Google Scholar] [CrossRef]
- Davis, R.; Nadin-Davis, S.A.; Moore, M.; Hanlon, C. Genetic characterization and phylogenetic analysis of skunk-associated rabies viruses in North America with special emphasis on the central plains. Virus Res. 2013, 174, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Sabeta, C.T.; Shumba, W.; Mohale, D.K.; Miyen, J.M.; Wandeler, A.I.; Nel, L.H. Mongoose rabies and the African civet in Zimbabwe. Vet. Rec. 2008, 163, 580. [Google Scholar] [CrossRef]
- Dah, I.; Poueme Namegni, R.S.; Mohamed, M.M.M.; Jumbo, S.D.; Noumedem, R.N.G.; Conclois, I.; Florian, L.; God-Yang, L.; Kameni, J.M.F.; Wade, A.; et al. Prevalence and public health significance of Lyssavirus in bats in North region of Cameroon. bioRxiv 2022. [Google Scholar] [CrossRef]
- Jacquot, M.; Wallace, M.A.; Streicker, D.G.; Biek, R. Geographic Range Overlap Rather than Phylogenetic Distance Explains Rabies Virus Transmission among Closely Related Bat Species. Viruses 2022, 14, 2399. [Google Scholar] [CrossRef] [PubMed]
- Lorente-Martínez, H.; Agorreta, A.; San Mauro, D. Genomic Fishing and Data Processing for Molecular Evolution Research. Methods Protoc. 2022, 5, 26. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zheng, B.; Jiang, X.; Chen, J.; Wang, S. Characteristics and Source of Dissolved Organic Matter in Lake Hulun, A Large Shallow Eutrophic Steppe Lake in Northern China. Water 2020, 12, 953. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Lambert, R.; Quiles, F.A.; Cabello-Díaz, J.M.; Piedras, P. Purification and identification of a nuclease activity in embryo axes from French bean. Plant Sci. Int. J. Exp. Plant Biol. 2014, 224, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Del Amparo, R.; Branco, C.; Arenas, J.; Vicens, A.; Arenas, M. Analysis of selection in protein-coding sequences accounting for common biases. Brief Bioinform. 2021, 22, bbaa431. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, Y.; Sun, G.; Wang, X.; Wu, X.; Gao, X.; Sha, W.; Yang, G.; Zhang, H. FGF gene family characterization provides insights into its adaptive evolution in Carnivora. Ecol. Evol. 2021, 11, 9837–9847. [Google Scholar] [CrossRef]
- Wu, X.; Chen, J.; Wang, X.; Shang, Y.; Wei, Q.; Zhang, H. Evolutionary Impacts of Pattern Recognition Receptor Genes on Carnivora Complex Habitat Stress Adaptation. Animals 2022, 12, 3331. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, J.L.; Dunn, K.A.; Mingrone, J.; Wood, B.A.; Karpinski, B.A.; Sherwood, C.C.; Wildman, D.E.; Maynard, T.M.; Bielawski, J.P. Functional Divergence of the Nuclear Receptor NR2C1 as a Modulator of Pluripotentiality During Hominid Evolution. Genetics 2016, 203, 905–922. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. TIG 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Price, S.A.; Bininda-Emonds, O.R.; Gittleman, J.L. A complete phylogeny of the whales, dolphins and even-toed hoofed mammals (Cetartiodactyla). Biol. Rev. Camb. Philos. Soc. 2005, 80, 445–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bininda-Emonds, O.R.; Gittleman, J.L.; Purvis, A. Building large trees by combining phylogenetic information: A complete phylogeny of the extant Carnivora (Mammalia). Biol. Rev. Camb. Philos. Soc. 1999, 74, 143–175. [Google Scholar] [CrossRef] [Green Version]
- Ibsen, M.S.; Gad, H.H.; Andersen, L.L.; Hornung, V.; Julkunen, I.; Sarkar, S.N.; Hartmann, R. Structural and functional analysis reveals that human OASL binds dsRNA to enhance RIG-I signaling. Nucleic Acids Res. 2015, 43, 5236–5248. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Yu, D.; Fan, Y.; Peng, L.; Wu, Y.; Yao, Y.-G. Loss of RIG-I leads to a functional replacement with MDA5 in the Chinese tree shrew. Proc. Natl. Acad. Sci. USA 2016, 113, 10950–10955. [Google Scholar] [CrossRef] [Green Version]
- Walter, A.M.; Kurps, J.; De Wit, H.; Schöning, S.; Toft-Bertelsen, T.L.; Lauks, J.; Ziomkiewicz, I.; Weiss, A.N.; Schulz, A.; Fischer von Mollard, G.; et al. The SNARE protein vti1a functions in dense-core vesicle biogenesis. EMBO J. 2014, 33, 1681–1697. [Google Scholar] [CrossRef]
- Yngvadottir, B.; Xue, Y.; Searle, S.; Hunt, S.; Delgado, M.; Morrison, J.; Whittaker, P.; Deloukas, P.; Tyler-Smith, C. A genome-wide survey of the prevalence and evolutionary forces acting on human nonsense SNPs. Am. J. Hum. Genet. 2009, 84, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Huelsmann, M.; Hecker, N.; Springer, M.S.; Gatesy, J.; Sharma, V.; Hiller, M. Genes lost during the transition from land to water in cetaceans highlight genomic changes associated with aquatic adaptations. Sci. Adv. 2019, 5, eaaw6671. [Google Scholar] [CrossRef] [Green Version]
- Albalat, R.; Cañestro, C. Evolution by gene loss. Nat. Rev. Genet. 2016, 17, 379–391. [Google Scholar] [CrossRef]
- Sharma, V.; Hecker, N.; Roscito, J.G.; Foerster, L.; Langer, B.E.; Hiller, M. Author Correction: A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nat. Commun. 2019, 10, 5707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, N.; Sharma, V.; Hiller, M. Convergent gene losses illuminate metabolic and physiological changes in herbivores and carnivores. Proc. Natl. Acad. Sci. USA 2019, 116, 3036–3041. [Google Scholar] [CrossRef] [Green Version]
- Choi, U.Y.; Kang, J.-S.; Hwang, Y.S.; Kim, Y.-J. Oligoadenylate synthase-like (OASL) proteins: Dual functions and associations with diseases. Exp. Mol. Med. 2015, 47, e144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Wang, X.; Xing, Y.; Rong, E.; Ning, M.; Smith, J.; Huang, Y. Origin and development of oligoadenylate synthetase immune system. BMC Evol. Biol. 2018, 18, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perelygin, A.A.; Zharkikh, A.A.; Scherbik, S.V.; Brinton, M.A. The Mammalian 2′-5′ Oligoadenylate Synthetase Gene Family: Evidence for Concerted Evolution of Paralogous Oas1 Genes in Rodentia and Artiodactyla. J. Mol. Evol. 2006, 63, 562–576. [Google Scholar] [CrossRef]
- Donovan, J.; Whitney, G.; Rath, S.; Korennykh, A. Structural mechanism of sensing long dsRNA via a noncatalytic domain in human oligoadenylate synthetase 3. Proc. Natl. Acad. Sci. USA 2015, 112, 3949–3954. [Google Scholar] [CrossRef] [Green Version]
- Carey, C.M.; Govande, A.A.; Cooper, J.M.; Hartley, M.K.; Kranzusch, P.J.; Elde, N.C. Recurrent Loss-of-Function Mutations Reveal Costs to OAS1 Antiviral Activity in Primates. Cell Host Microbe 2019, 25, 336–343.e334. [Google Scholar] [CrossRef]
- Luis, A.D.; Hayman, D.T.; O’Shea, T.J.; Cryan, P.M.; Gilbert, A.T.; Pulliam, J.R.; Mills, J.N.; Timonin, M.E.; Willis, C.K.; Cunningham, A.A.; et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proc. Biol. Sci. 2013, 280, 20122753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itan, Y.; Shang, L.; Boisson, B.; Patin, E.; Bolze, A.; Moncada-Vélez, M.; Scott, E.; Ciancanelli, M.J.; Lafaille, F.G.; Markle, J.G.; et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 13615–13620. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Banerjee, S.; Wang, Y.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Silverman, R.H.; Weiss, S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 2241–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.I.; Ernst, R.K.; Bader, M.W. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 2005, 3, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Van Valen, L. A New Evolutionary Law. In Evolutionary Theory; University of Chicago Press: Chicago, IL, USA, 1973. [Google Scholar]
- Lee, W.-B.; Choi, W.Y.; Lee, D.-H.; Shim, H.; Kim-Ha, J.; Kim, Y.-J. OAS1 and OAS3 negatively regulate the expression of chemokines and interferon-responsive genes in human macrophages. BMB Rep. 2019, 52, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Tian, L.; Wang, J.; Zheng, B.; Zhang, W. EV71 3C protease cleaves host anti-viral factor OAS3 and enhances virus replication. Virol. Sin. 2022, 37, 418–426. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, S.; Xu, J.; Chen, B.; Zhou, K.; Yang, G. Phylogenomic analysis resolves the interordinal relationships and rapid diversification of the laurasiatherian mammals. Syst. Biol. 2012, 61, 150–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, M.B.; Deak, T.; Schiml, P.A. Sociality and sickness: Have cytokines evolved to serve social functions beyond times of pathogen exposure? Brain Behav. Immun. 2014, 37, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Hill, T.; Koseva, B.S.; Unckless, R.L. The Genome of Drosophila innubila Reveals Lineage-Specific Patterns of Selection in Immune Genes. Mol. Biol. Evol. 2019, 36, 1405–1417. [Google Scholar] [CrossRef]
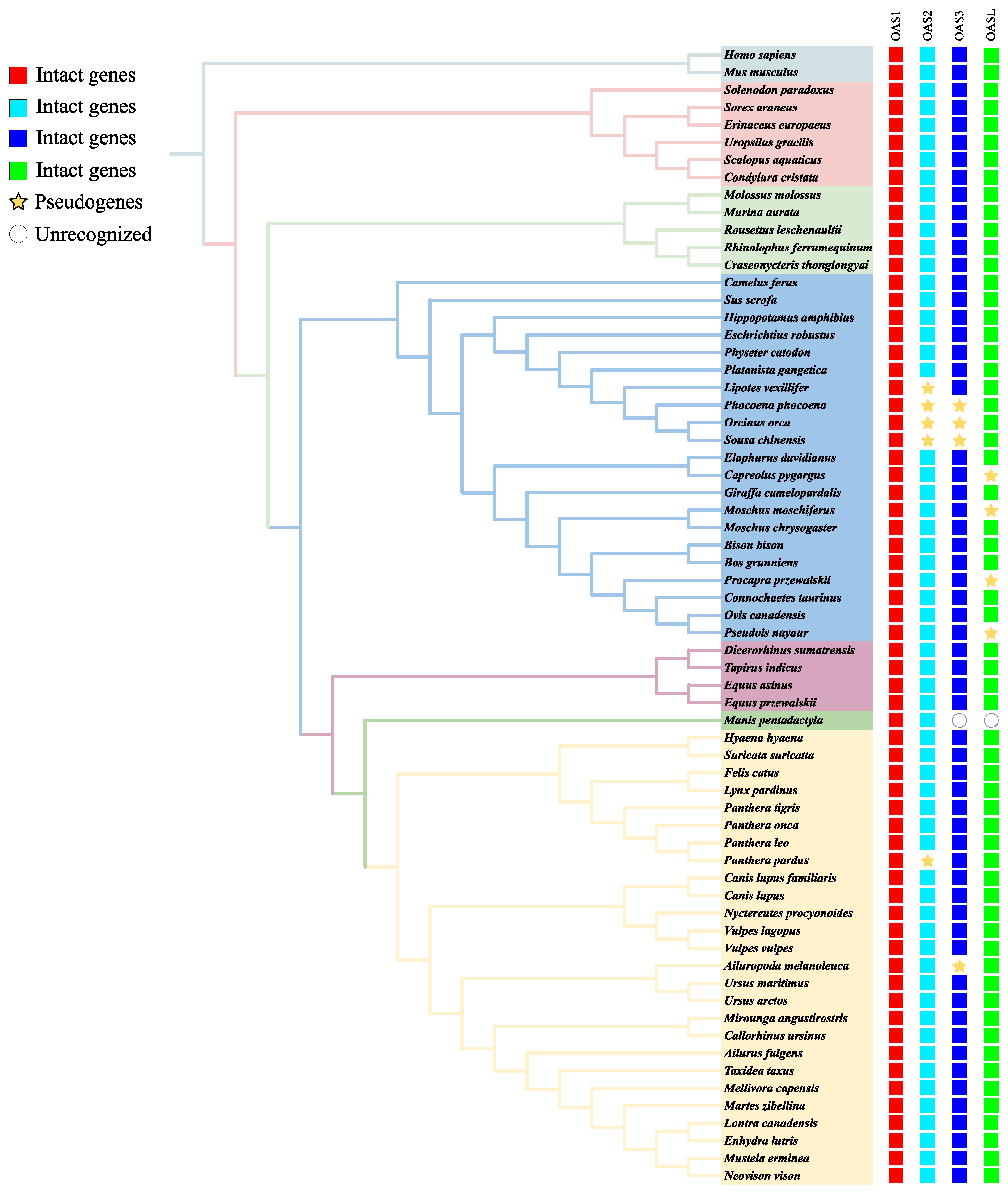

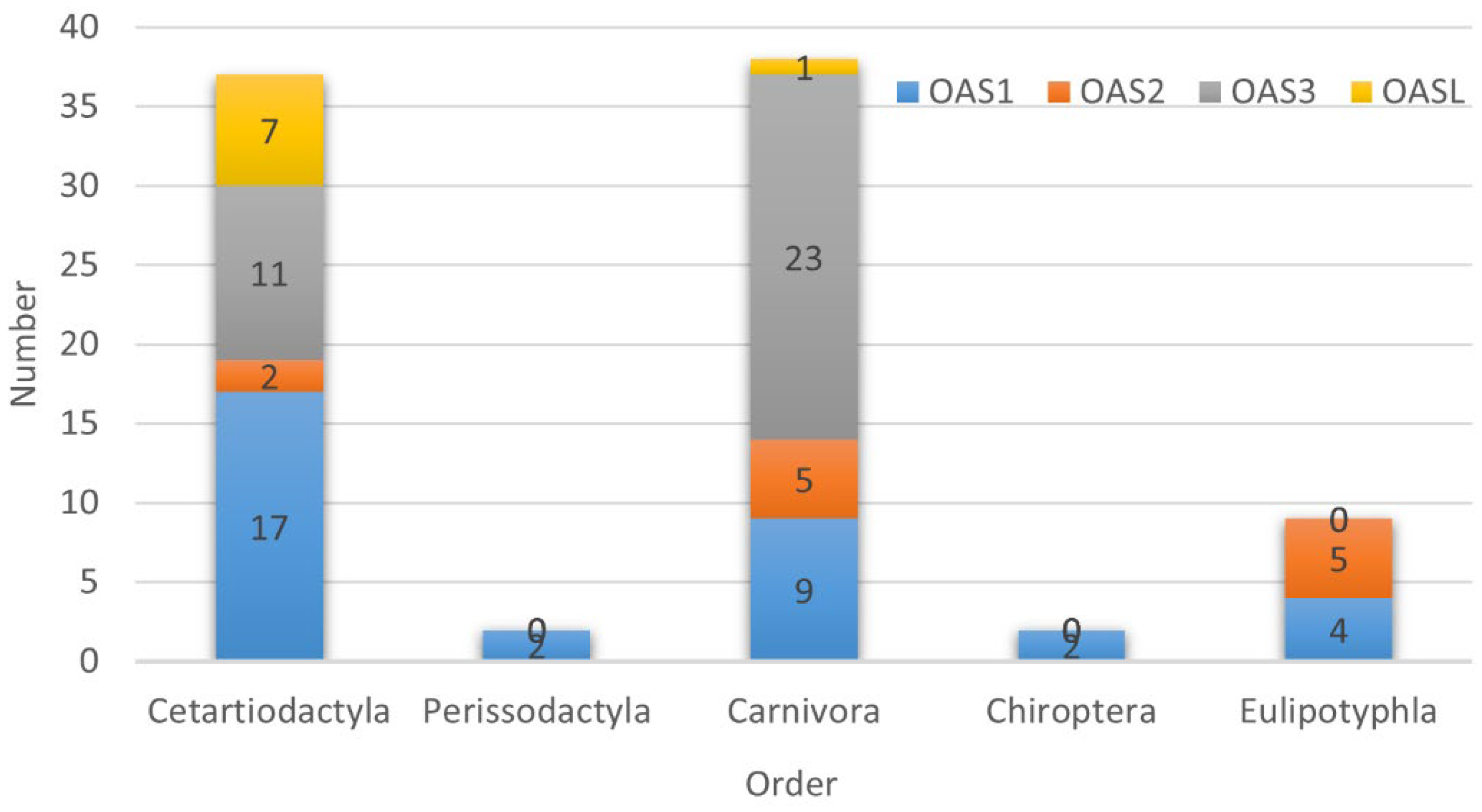
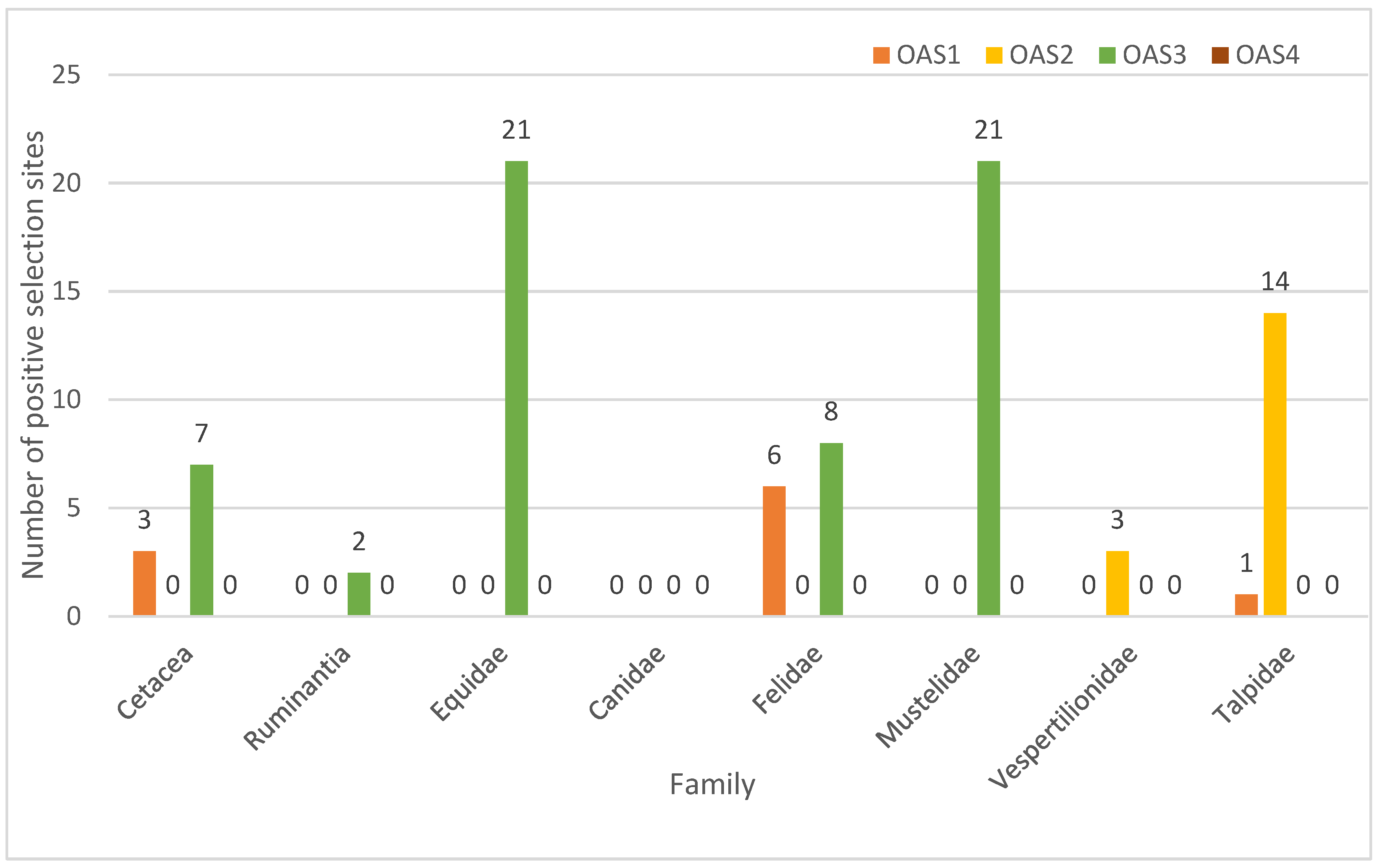

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Gene | lnL M7 | lnL M8 | No. of Species | df | p-Values | BEB |
|---|---|---|---|---|---|---|---|
| Cetartiodactyla | OAS1 | −6072.4755 | −6032.329 | 21 | 2 | 3.67 × 10−18 ** | 9 K 0.999 ** 10 S 0.998 ** 26 R 0.967 * 47 C 1.000 ** 110 T 0.951 * 136 A 0.997 ** 189 N 0.999 ** 206 L 0.998 ** 234 K 0.965 * 237 R 0.994 ** 239 N 0.968 * 256 K 0.987 * 263 K 0.975 * 277 T 0.977 * 280 A 0.965 * 302 Y 0.996 ** 312 L 0.969 * |
| OAS2 | −9839.0114 | −9832.432 | 17 | 2 | 4.41 × 10−5 ** | 164 H 0.968 * 275 D 0.967 * | |
| OAS3 | −7911.3451 | −7900.636 | 17 | 2 | 1.07 × 10−30 ** | 7 R 0.999 ** 8 C 1.000 * 11 V 1.000 * 255 G 0.982 * 256 R 0.998 ** 268 G 1.000 ** 275 A 0.999 ** 276 S 1.000 ** 280 L 0.999 ** 468 N 0.954 * 553 R 0.997 ** | |
| OASL | −9371.3533 | −9371.353 | 17 | 2 | 2.05 × 10−14** | 106 S 0.996 ** 120 S 0.979 * 143 S 0.985 * 147 F 0.998 ** 160 R 1.000 ** 234 R 0.961 * 241 R 0.984 * | |
| Pholidota | OAS1 | −10,065.963 | −10,065.963 | −2E-06 | 2 | 0.5 | |
| OAS2 | −12,224.198 | −12,224.198 | 0 | 2 | 0.5 | ||
| Perissodactyla | OAS1 | −2693.1406 | −2685.5063 | 4 | 2 | 0.000483 ** | 375 P 0.973 * 378 T 0.995 ** |
| OAS2 | −4982.3562 | −4983.0484 | 4 | 2 | 0.500476317 | ||
| OAS3 | −7739.7452 | −7740.2327 | 4 | 2 | 0.614127 | ||
| OASL | −1712.6166 | −1715.0368 | 4 | 2 | 0.5 | ||
| Carnivora | OAS1 | −7562.7926 | −7593.4765 | 26 | 2 | 4.72 × 10−14 ** | 11 Y 0.980 * 158 G 0.987 * 161 N 0.969 * 163 E 0.982 * 167 T 0.995 ** 170 R 0.997 ** 173 Q 0.978 * 189 Q 0.978 * 273 G 0.992 ** |
| OAS2 | −11,635.337 | −11,649.541 | 25 | 2 | 6.77 × 10−7 ** | 188 H 0.985 * 213 L 0.965 * 215 H 0.989 * 368 E 0.984 * 376 R 0.971 * | |
| OAS3 | −20,393.582 | −20,496.004 | 25 | 2 | 3.30 × 10−45 ** | 1 D 0.990 ** 2 S 0.997 ** 4 V 0.999 ** 6 R 0.996 ** 7 N 0.998 ** 8 L 0.986 ** 9 M 0.980 * 11 S 0.999 ** 13 L 0.993 ** 14 A 0.959 * 15 A 0.999 ** 24 A 0.997 ** 36 K 0.995 ** 69 R 0.996 ** 92 H 0.961 * 136 S 0.995 * 172 T 0.987 * 189 C 1.000 ** 260 R 0.988 * 335 S 0.998 ** 348 T 0.978 * 407 S 0.996 ** 520 S 0.980 * | |
| OASL | −7234.9379 | −7242.5053 | 26 | 2 | 0.0005169 ** | 390 H 0.977 * | |
| Chiroptera | OAS1 | −4036.7745 | −4047.5839 | 5 | 2 | 2.02 × 10−5 ** | 4 E 0.954 * 231 T 0.968 * |
| OAS2 | −3866.3837 | −3866.5936 | 5 | 2 | 0.810632 | ||
| OAS3 | −10,106.391 | −10,113.077 | 5 | 2 | 0.0012473 ** | ||
| OASL | −6080.1380 | −6084.3458 | 5 | 2 | 0.0148787 ** | ||
| Eulipotyphla | OAS1 | −4114.7990 | −4124.5812 | 6 | 2 | 5.64 × 10−5 ** | 153 L 0.95 9 * 154 R 0.977 * 212 P 0.967 * 234 D 0.966 * |
| OAS2 | −6875.2726 | −6896.9948 | 6 | 2 | 3.68 × 10−10 ** | 144 K 0.962 * 162 E 0.988 * 189 S 0.980 * 253 N 0.975 * 310 R 0.979 * | |
| OAS3 | −12,026.957 | −12,033.766 | 6 | 2 | 0.0011045 * | ||
| OASL | −2818.2825 | −2821.8173 | 6 | 2 | 0.02916485 * |
| Taxa | Gene | Positive Selection (0.9) | Positive Selection (0.95) |
|---|---|---|---|
| Cetartiodactyla | OAS1 | 27 | 14 |
| OAS2 | 4 | 1 | |
| OAS3 | 6 | 1 | |
| OASL | 6 | 1 | |
| Perissodactyla | OAS1 | 1 | 1 |
| OAS2 | 4 | 0 | |
| OAS3 | 0 | 0 | |
| OASL | 3 | 1 | |
| Carnivora | OAS1 | 1 | 0 |
| OAS2 | 1 | 1 | |
| OAS3 | 2 | 0 | |
| OASL | 1 | 1 | |
| Chiroptera | OAS1 | 4 | 1 |
| OAS2 | 2 | 0 | |
| OAS3 | 2 | 0 | |
| OASL | 0 | 0 | |
| Eulipotyphla | OAS1 | 1 | 0 |
| OAS2 | 3 | 0 | |
| OAS3 | 2 | 0 | |
| OASL | 3 | 0 |
| Taxa | Gene | lnL M0 | lnL M1 | p-Values | ω0 | ω | Order |
|---|---|---|---|---|---|---|---|
| Carnivora | OAS1 | −7886.75 | −7885.87 | 0.9393 | 0.37587 | 0.42983 0.32010 0.35686 0.32953 0.41482 0.29480 | Mustelidae Canidae Feliformia Ailuridae pinnipedia Ursidae |
| OAS2 | −11,883.4 | −11,867.53 | 1.75E-05 ** | 0.33957 | 0.44169 0.50012 0.30951 0.36258 0.55048 0.20961 | Mustelidae Canidae Feliformia Ailuridae pinnipedia Ursidae | |
| OAS3 | −21,140.7 | −21,130.42 | 0.00218 ** | 0.31004 | 0.23408 0.46858 0.33034 0.34092 0.34686 0.83956 | Mustelidae Canidae Feliformia Ailuridae pinnipedia Ursidae | |
| OASL | −7315.46 | −7311.68 | 0.27016 | 0.3606 | 0.34322 0.37139 0.26894 0.49875 0.60788 0.40809 | Mustelidae Canidae Feliformia Ailuridae pinnipedia Ursidae | |
| Eulipotyphla | OAS1 | −4257.98 | −4256.004 | 0.4122 | 0.2864 | 0.29963 0.32943 0.32723 0.18820 | Talpidae Erinaceidae Solenodontidae Soricidea |
| OAS2 | −7181.19 | −7140.64 | 1.01E-16 ** | 0.20656 | 0.45990 0.11458 0.16019 0.03739 | Talpidae Erinaceidae Solenodontidae Soricidea | |
| OAS3 | −12,355.6 | −12,349.08 | 0.01026 * | 0.19660 | 0.19437 0.16947 0.20071 0.14918 | Talpidae Erinaceidae Solenodontidae Soricidea | |
| OASL | −2867.49 | −2863.16 | 0.0737 | 0.34407 | 0.29138 0.34153 0.77844 0.26496 | Talpidae Erinaceidae Solenodontidae Soricidea | |
| Chiroptera | OAS1 | −4183.03 | −4164.62 | 6.48E-07 ** | 0.24726 | 0.27718 0.35271 0.38371 0.27003 0.00970 | Rhinolophidae Vespertilionidae Molossidae Pteropodidae Nycteridae |
| OAS2 | −3940.66 | −3937.56 | 0.2871 | 2.48390 | 0.15611 0.14157 0.14517 0.12003 0.50989 | Rhinolophidae Vespertilionidae Molossidae Pteropodidae Nycteridae | |
| OAS3 | −10,250.7 | −10,247.75 | 0.3047 | 0.30613 | 0.23936 0.33791 0.26711 0.35137 0.30723 | Rhinolophidae Vespertilionidae Molossidae Pteropodidae Nycteridae | |
| OASL | −6204.10 | −6193.65 | 0.3047 | 0.27926 | 0.23185 0.31819 0.43633 0.17294 0.05074 | Rhinolophidae Vespertilionidae Molossidae Pteropodidae Nycteridae | |
| Cetartiodactyla | OAS1 | −6270.46 | −6264.38 | 0.14158 | 0.49856 | 0.27123 0.52040 0.28835 0.31889 0.84189 0.65807 0.37722 0.41818 | Moschidae Bovidae Cervidae Giraffidae Hippopotamidae Odontoceti Camelidae Suidae |
| OAS2 | −4926.82 | −4921.97 | 0.258364 | 0.467758 | 0.66480 0.52956 0.74130 0.44746 0.48362 0.66494 0.45639 0.33798 | Moschidae Bovidae Cervidae Giraffidae Hippopotamidae Odontoceti Camelidae Suidae | |
| OAS3 | −8328.28 | −8317.61 | 1.32E-05 ** | 0.426209 | 0.26979 0.59390 0.38201 0.80675 0.50011 0.36829 0.31322 0.32099 | Moschidae Bovidae Cervidae Giraffidae Hippopotamidae Odontoceti Camelidae Suidae | |
| OASL | −5154.22 | −5126.18 | 2.72E-09 ** | 0.50055 | 0.02728 0.71554 1.02181 1.03467 0.71635 0.40102 0.46758 0.67712 | Moschidae Bovidae Cervidae Giraffidae Hippopotamidae Odontoceti Camelidae Suidae | |
| Perissodactyla | OAS1 | −2705.67 | −2699.96 | 0.0097 ** | 0.37987 | 0.68667 0.63466 0.44241 | Equidae Rhinocerotidae Tapiridae |
| OAS2 | −5009.23 | −5007.79 | 0.4109 | 0.40647 | 0.22805 0.45730 0.32208 | Equidae Rhinocerotidae Tapiridae | |
| OAS3 | −7799.66 | −7796.08 | 0.0669 | 0.2828 | 0.37966 0.29006 0.27405 | Equidae Rhinocerotidae Tapiridae | |
| OASL | −1722.41 | −1722.147 | 0.9124 | 0.2445 | 0.31200 0.26917 0.19742 | Equidae Rhinocerotidae Tapiridae |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Wu, X.; Shang, Y.; Wang, X.; Zhou, S.; Zhang, H. Adaptive Evolution of the OAS Gene Family Provides New Insights into the Antiviral Ability of Laurasiatherian Mammals. Animals 2023, 13, 209. https://doi.org/10.3390/ani13020209
Liu G, Wu X, Shang Y, Wang X, Zhou S, Zhang H. Adaptive Evolution of the OAS Gene Family Provides New Insights into the Antiviral Ability of Laurasiatherian Mammals. Animals. 2023; 13(2):209. https://doi.org/10.3390/ani13020209
Chicago/Turabian StyleLiu, Gang, Xiaoyang Wu, Yongquan Shang, Xibao Wang, Shengyang Zhou, and Honghai Zhang. 2023. "Adaptive Evolution of the OAS Gene Family Provides New Insights into the Antiviral Ability of Laurasiatherian Mammals" Animals 13, no. 2: 209. https://doi.org/10.3390/ani13020209