
The Hoof Color of Australian White Sheep Is Associated with Genetic Variation of the MITF Gene
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Hoof Color Data Collection
2.2. Whole Genome Sequencing, Genotyping and Quality Control
2.3. GWAS of Hoof Color Traits in Australian White Sheep
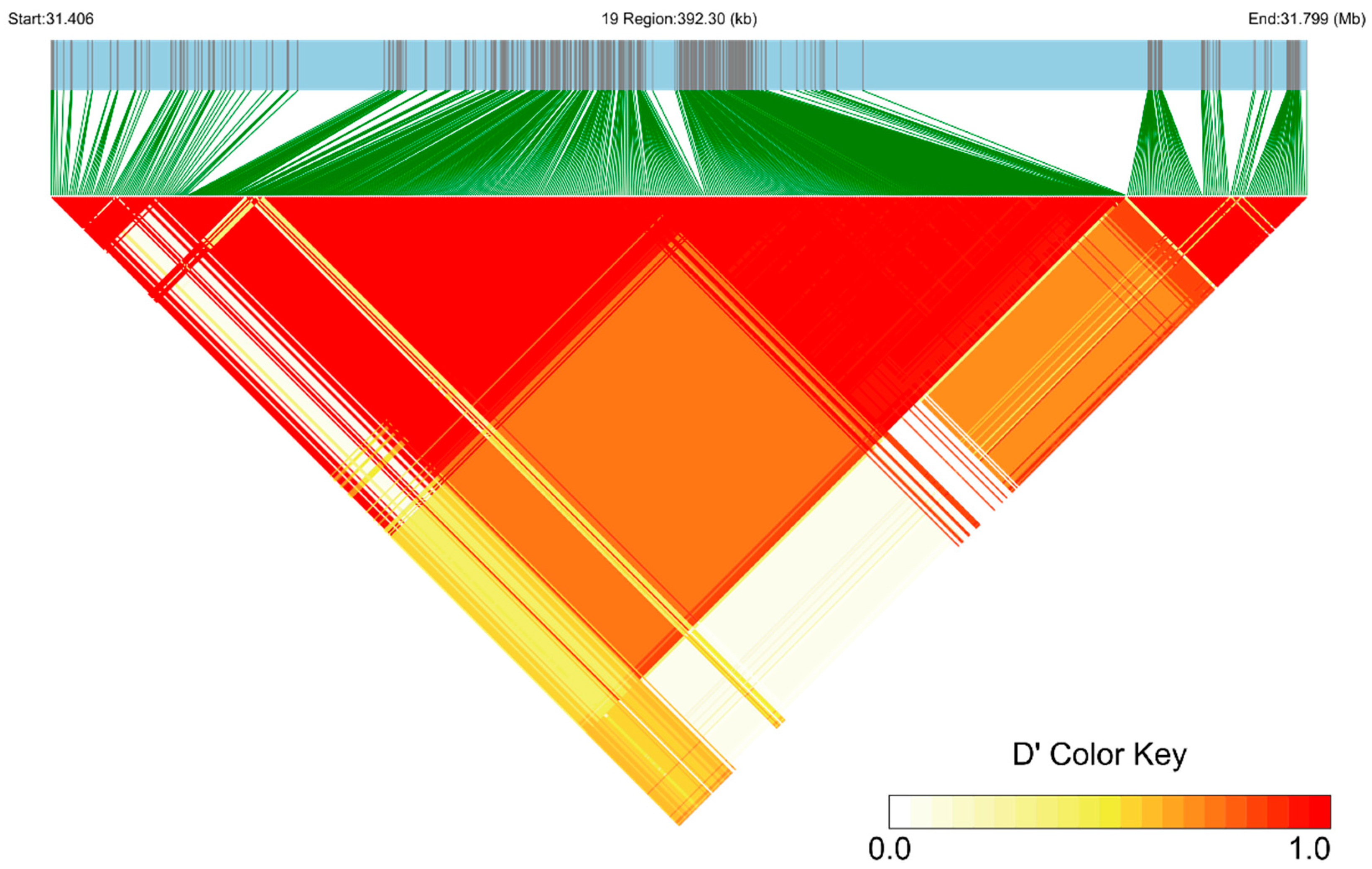
2.4. Linkage Disequilibrium and Haplotype Analysis
2.5. Validation of Selected SNP in Six Australian White Sheep
2.6. Association between the Hoof Color and Different SNP Genotypes in a Validation Population of 212 AUW Sheep
2.7. Bioinformatics Analysis
3. Results
3.1. Descriptive Statistics
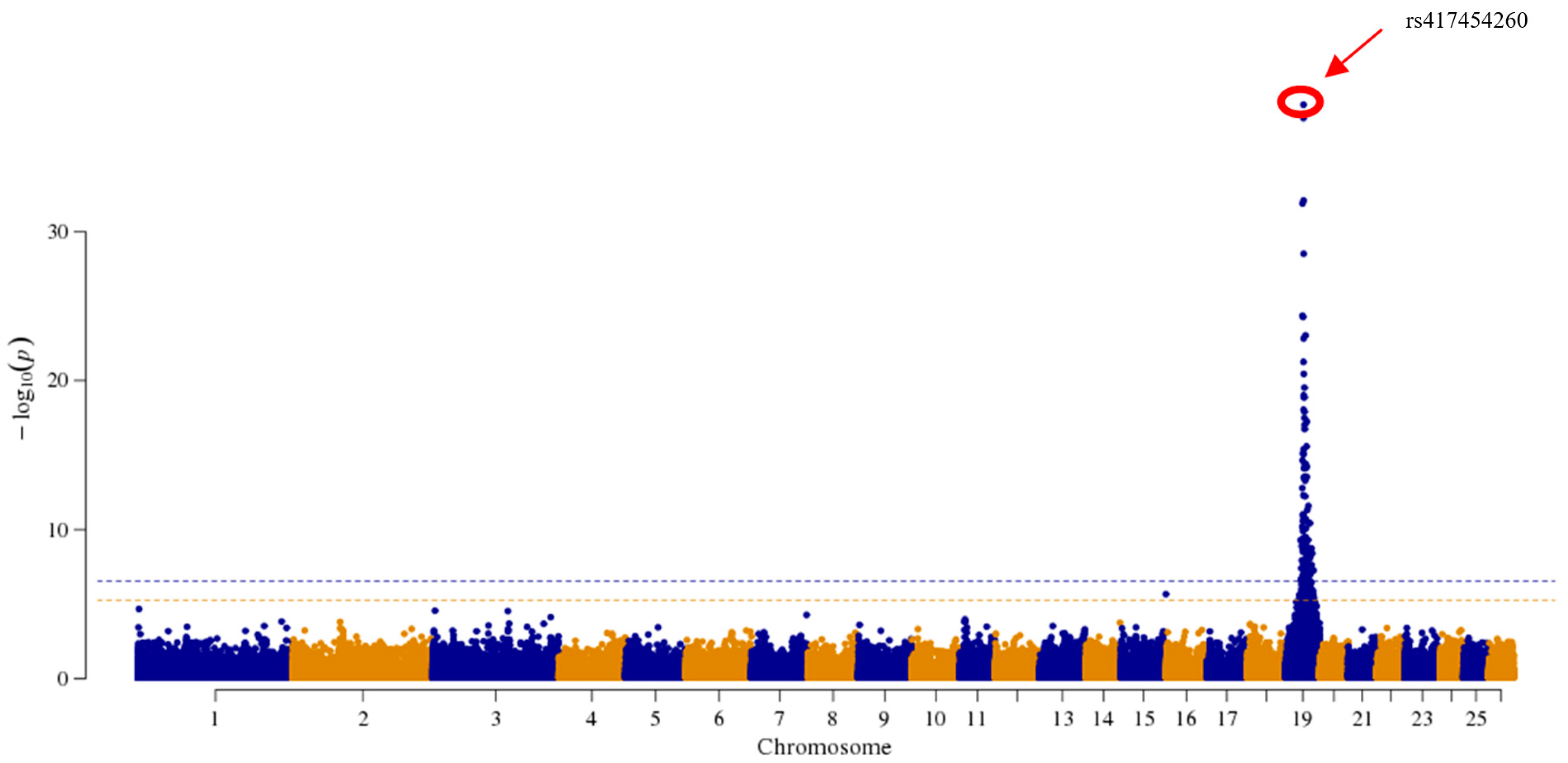
3.2. Genome-Wide Association Study
3.3. Validation of SNP Loci
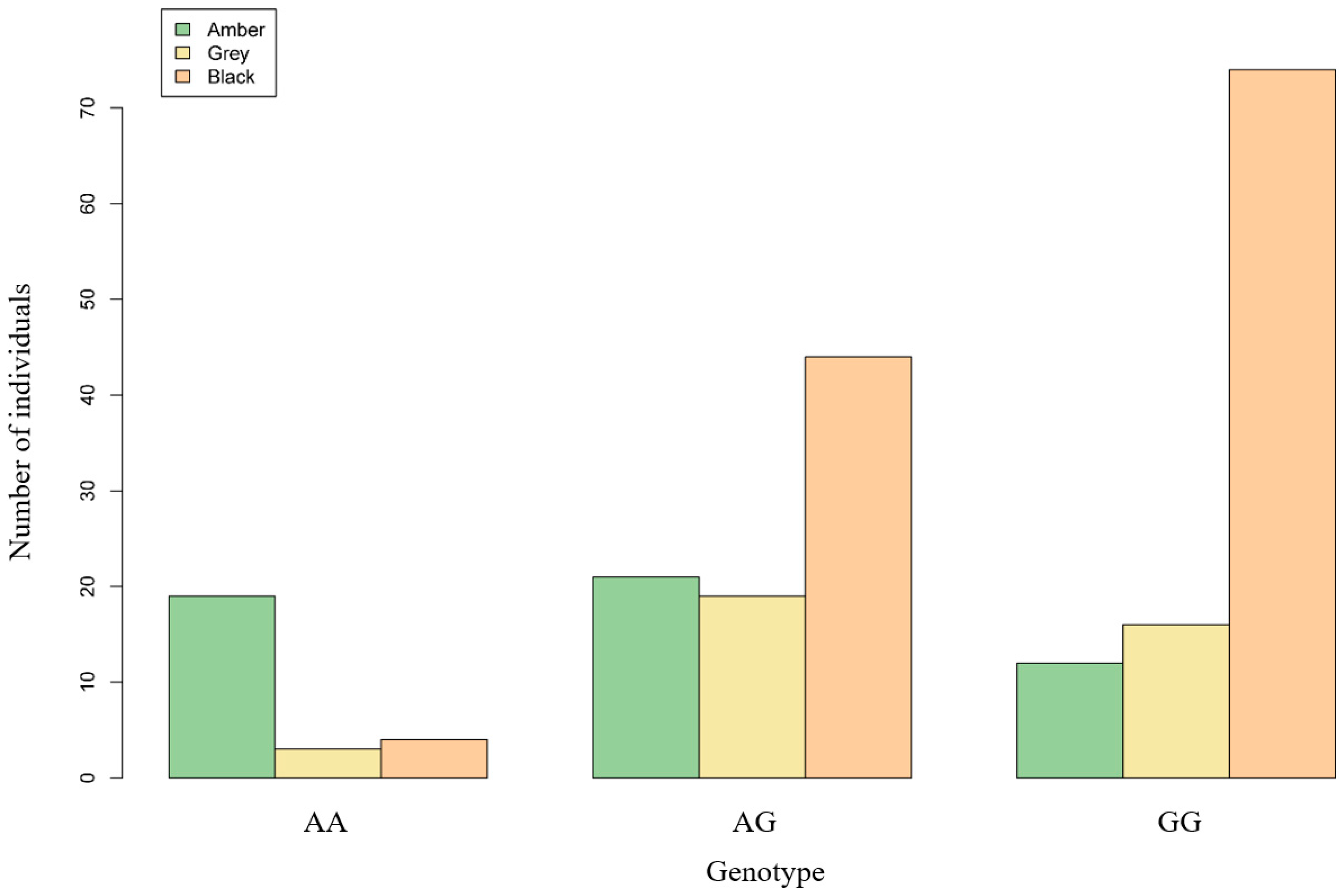
3.4. Association Analysis of the Locus in the MITF Gene
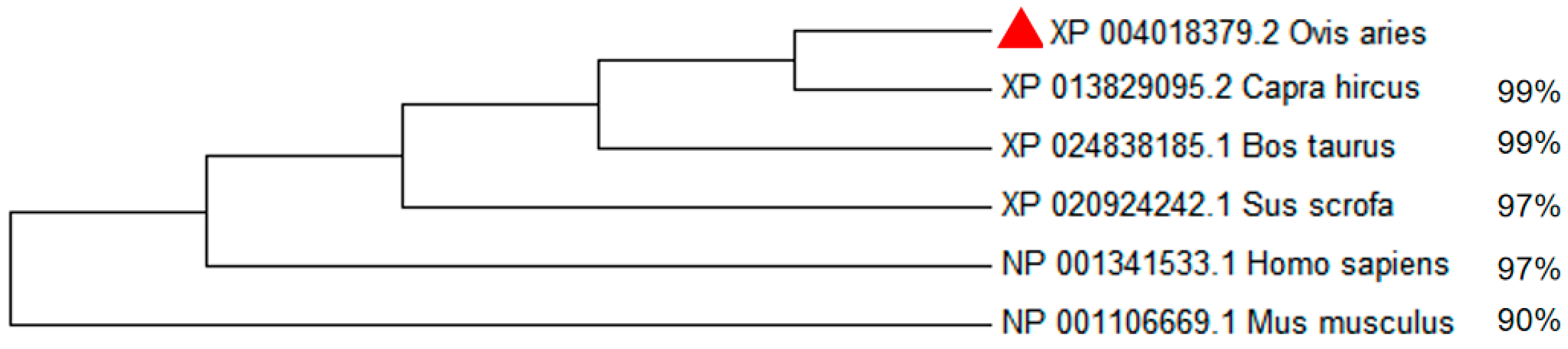
3.5. Construction of Phylogenetic Tree of MITF Gene in Six Species and Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klungland, H.; Vage, D. Pigmentary switches in domestic animal species. Ann. N. Y. Acad. Sci. 2003, 994, 331–338. [Google Scholar] [CrossRef]
- Xu, X.; Dong, G.; Schmidt-Küntzel, A.; Zhang, X.; Zhuang, Y.; Fang, R.; Sun, X.; Hu, X.; Zhang, T.; Yang, H.; et al. The genetics of tiger pelage color variations. Cell Res. 2017, 27, 954–957. [Google Scholar] [CrossRef]
- Protas, M.; Patel, N. Evolution of coloration patterns. Annu. Rev. Cell Dev. Biol. 2008, 24, 425–446. [Google Scholar] [CrossRef]
- Royo, L.; Alvarez, I.; Arranz, J.; Fernández, I.; Rodríguez, A.; Pérez-Pardal, L.; Goyache, F. Differences in the expression of the ASIP gene are involved in the recessive black coat colour pattern in sheep: Evidence from the rare Xalda sheep breed. Anim. Genet. 2008, 39, 290–293. [Google Scholar] [CrossRef]
- Fontanesi, L.; Dall’Olio, S.; Beretti, F.; Portolano, B.; Russo, V. Coat colours in the Massese sheep breed are associated with mutations in the agouti signalling protein (ASIP) and melanocortin 1 receptor (MC1R) genes. Animal 2011, 5, 8–17. [Google Scholar] [CrossRef]
- Cruz, A.; Yucra, A.; Gutiérrez, G.; Burgos, A.; Morante, R.; Gutiérrez, J.; Cervantes, I.; Wurzinger, M. Colorimetry analysis of coat color and its relationship with fiber traits in alpacas. Animal 2021, 15, 100219. [Google Scholar] [CrossRef]
- Chang, T. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef]
- Chen, W.; Tseng, T.; Hsiao, N.; Lin, Y.; Wen, Z.; Tsai, C.; Lee, Y.; Lin, H.; Tsai, K. Discovery of highly potent tyrosinase inhibitor, T1, with significant anti-melanogenesis ability by zebrafish in vivo assay and computational molecular modeling. Sci. Rep. 2015, 5, 7995. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J. Research progress on molecular mechanism in the formation of melanin. J. Xinjiang Univ. 2019, 36, 468–474+499. [Google Scholar]
- Vandamme, N.; Berx, G. From neural crest cells to melanocytes: Cellular plasticity during development and beyond. Cell Mol. Life Sci. 2019, 76, 1919–1934. [Google Scholar] [CrossRef]
- Dong, C.; Wang, H.; Xue, L.; Dong, Y.; Yang, L.; Fan, R.; Yu, X.; Tian, X.; Ma, S.; Smith, G. Coat color determination by miR-137 mediated down-regulation of microphthalmia-associated transcription factor in a mouse model. RNA 2012, 18, 1679–1686. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, A.; Estellé, J.; Blin, A.; Muñoz, M.; Créchet, F.; Demenais, F.; Vincent-Naulleau, S.; Bourneuf, E. KIT and melanoma predisposition in pigs: Sequence variants and association analysis. Anim. Genet. 2014, 45, 445–458. [Google Scholar] [CrossRef]
- Kim, K.; Lee, T.; Cho, E. SH3BP4, a novel pigmentation gene, is inversely regulated by miR-125b and MITF. Exp Mol. Med. 2017, 49, e367. [Google Scholar] [CrossRef]
- Zhou, Q.; Cao, C.; Zhang, H.; Liang, Y.; Zhang, X.; Kang, Y.; Fang, W.; Lan, X.; Li, R.; Pan, C. Convergent changes in melanocortin receptor 1 gene are associated with black-headed coat color in sheep. J. Anim. Sci. 2023, 101, skad084. [Google Scholar] [CrossRef]
- Brancalion, L.; Haase, B.; Wade, C. Canine coat pigmentation genetics: A review. Anim. Genet. 2022, 53, 3–34. [Google Scholar] [CrossRef]
- Klein, R.; Zeiss, C.; Chew, E.; Tsai, J.; Sackler, R.; Haynes, C.; Henning, A.; SanGiovanni, J.; Mane, S.; Mayne, S.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Yengo, L.; Vedantam, S.; Marouli, E.; Sidorenko, J.; Bartell, E.; Sakaue, S.; Graff, M.; Eliasen, A.; Jiang, Y.; Raghavan, S.; et al. A saturated map of common genetic variants associated with human height. Nature 2022, 610, 704–712. [Google Scholar] [CrossRef]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.; Chen, T.; Wang, Q.; Bolla, M.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef]
- Evangelou, E.; Warren, H.; Mosen-Ansorena, D.; Mifsud, B.; Pazoki, R.; Gao, H.; Ntritsos, G.; Dimou, N.; Cabrera, C.; Karaman, I.; et al. Million Veteran Program. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018, 50, 1412–1425. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Shen, M.; Xie, X.; Liu, G.; Xu, Y.; Lv, F.; Yang, H.; Yang, Y.; Liu, C.; et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Liu, W.; Li, Y.; Pan, J.; Pu, Y.; Han, J.; Orlando, L.; Ma, Y.; Jiang, L. A single-nucleotide mutation within the TBX3 enhancer increased body size in Chinese horses. Curr. Biol. 2022, 32, 480–487.e6. [Google Scholar] [CrossRef]
- Li, Y.; Gong, Y.; Zhang, Z.; Li, L.; Liu, X.; He, X.; Zhao, Q.; Pu, Y.; Ma, Y.; Jiang, L. Whole-genome sequencing reveals selection signals among Chinese, Pakistani, and Nepalese goats. J. Genet. Genom. 2023, 50, 362–365. [Google Scholar] [CrossRef]
- Voß, K.; Blaj, I.; Tetens, J.; Thaller, G.; Becker, D. Roan coat color in livestock. Anim. Genet. 2022, 53, 549–556. [Google Scholar] [CrossRef]
- Bellone, R. Pleiotropic effects of pigmentation genes in horses. Anim. Genet. 2010, 41 (Suppl. S2), 100–110. [Google Scholar] [CrossRef]
- Andersson, L.; Archibald, A.; Bottema, C.; Brauning, R.; Burgess, S.; Burt, D.; Casas, E.; Cheng, H.; Clarke, L.; Couldrey, C.; et al. FAANG Consortium. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 2015, 16, 57. [Google Scholar] [CrossRef]
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.; Pairo-Castineira, E.; et al. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 4, 1438–1447. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, S.; Xia, C.; Gao, Y.; Pan, Z.; Canela-Xandri, O.; Khamseh, A.; Rawlik, K.; Wang, S.; Li, B.; et al. Comparative transcriptome in large-scale human and cattle populations. Genome Biol. 2022, 23, 176. [Google Scholar] [CrossRef]
- Pan, Z.; Yao, Y.; Yin, H.; Cai, Z.; Wang, Y.; Bai, L.; Kern, C.; Halstead, M.; Chanthavixay, G.; Trakooljul, N.; et al. Pig genome functional annotation enhances the biological interpretation of complex traits and human disease. Nat. Commun. 2021, 12, 5848. [Google Scholar] [CrossRef]
- Li, H.; Xu, H.; Akhatayeva, Z.; Liu, H.; Lin, C.; Han, X.; Lu, X.; Lan, X.; Zhang, Q.; Pan, C. Novel indel variations of the sheep FecB gene and their effects on litter size. Gene 2021, 767, 145176. [Google Scholar] [CrossRef]
- McCormick, R.; Truong, S.; Mullet, J. RIG: Recalibration and interrelation of genomic sequence data with the GATK. G3 Genes Genomes Genet. 2015, 5, 655–665. [Google Scholar] [CrossRef]
- Song, S.; Yao, N.; Yang, M.; Liu, X.; Dong, K.; Zhao, Q.; Pu, Y.; He, X.; Guan, W.; Yang, N.; et al. Exome sequencing reveals genetic differentiation due to high-altitude adaptation in the Tibetan cashmere goat (Capra hircus). BMC Genom. 2016, 17, 122. [Google Scholar] [CrossRef]
- Ghasemi, M.; Zamani, P.; Vatankhah, M.; Abdoli, R. Genome-wide association study of birth weight in sheep. Animal 2019, 13, 1797–1803. [Google Scholar] [CrossRef]
- Su, P.; Luo, Y.; Huang, Y.; Akhatayeva, Z.; Xin, D.; Guo, Z.; Pan, C.; Zhang, Q.; Xu, H.; Lan, X. Short variation of the sheep PDGFD gene is correlated with litter size. Gene 2022, 844, 146797. [Google Scholar] [CrossRef]
- Zheng, K.; Su, X.; Zheng, X.; Zhang, L.; Chen, Y.; Wu, K.; Li, T.; Zhang, Z.; Zhao, Z. First report of Stenotrophomonas maltophilia causing root soft rot of Sanqi (Panax notoginseng) in China. Plant Dis. 2021, 106, 755. [Google Scholar] [CrossRef]
- Fu, W.; Wang, R.; Nanaei, H.; Wang, J.; Hu, D.; Jiang, Y. RGD v2.0: A major update of the ruminant functional and evolutionary genomics database. Nucleic Acids Res. 2022, 50, D1091–D1099. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, P.; Fu, W.; Dong, T.; Qi, C.; Liu, L.; Guo, G.; Li, C.; Ciu, X.; Zhang, S.; et al. Genome-wide association study for pigmentation traits in Chinese Holstein population. Anim. Genet. 2014, 45, 740–744. [Google Scholar] [CrossRef]
- Puckett, E.; Davis, I.; Harper, D.; Wakamatsu, K.; Battu, G.; Belant, J.; Beyer, D.; Carpenter, C.; Crupi, A.; Davidson, M.; et al. Genetic architecture and evolution of color variation in American black bears. Curr. Biol. 2023, 33, 86–97.e10. [Google Scholar] [CrossRef]
- Leite, J.; Silva, R.; Asensio, L.; Sousa, J.; Silva, W.; Façanha, D. Coat color and morphological hair traits influence on the mechanisms related to the heat tolerance in hair sheep. Int. J. Biometeorol. 2020, 64, 2185–2194. [Google Scholar] [CrossRef]
- Wang, C.; Li, H.; Guo, Y.; Huang, J.; Sun, Y.; Min, J.; Wang, J.; Fang, X.; Zhao, Z.; Wang, S.; et al. Donkey genomes provide new insights into domestication and selection for coat color. Nat. Commun. 2020, 11, 6014. [Google Scholar] [CrossRef]
- Muniz, M.; Caetano, A.; McManus, C.; Cavalcanti, L.; Façanha, D.; Leite, J.; Facò, O.; Paiva, S. Application of genomic data to assist a community- based breeding program: A preliminary study of coat color genetics in Morada Nova sheep. Livest. Sci. 2016, 190, 89–93. [Google Scholar] [CrossRef]
- Li, M.; Tiirikka, T.; Kantanen, J. A genome-wide scan study identifies a single nucleotide substitution in ASIP associated with white versus non-white coat-colour variation in sheep (Ovis aries). Heredity 2014, 112, 122–131. [Google Scholar] [CrossRef]
- Kijas, J.; Serrano, M.; McCulloch, R.; Li, Y.; Ortiz, J.S.; Calvo, J.; Pérez-Guzmán, M. International Sheep Genomics Consortium. Genomewide association for a dominant pigmentation gene in sheep. J. Anim. Breed Genet. 2013, 130, 468–475. [Google Scholar] [CrossRef]
- Liu, L.; Harris, B.; Keehan, M.; Zhang, Y. Genome scan of pigmentation traits in Friesian-Jersey crossbred cattle. J Genet Genom. 2009, 36, 661–666. [Google Scholar] [CrossRef]
- Liang, D.; Zhao, P.; Si, J.; Fang, L.; Pairo-Castineira, E.; Hu, X.; Xu, Q.; Hou, Y.; Gong, Y.; Liang, Z.; et al. Genomic Analysis Revealed a Convergent Evolution of LINE-1 in Coat Color: A Case Study in Water Buffaloes (Bubalus bubalis). Mol. Biol. Evol. 2021, 38, 1122–1136. [Google Scholar] [CrossRef]
- Xu, X.; Dong, G.; Hu, X.; Miao, L.; Zhang, X.; Zhang, D.; Yang, H.; Zhang, T.; Zou, Z.; Zhang, T.; et al. The genetic basis of white tigers. Curr. Biol. 2013, 23, 1031–1045. [Google Scholar] [CrossRef]
- Caro, T.; Mallarino, R. Coloration in Mammals. Trends Ecol. Evol. 2020, 35, 357–366. [Google Scholar] [CrossRef]
- Goding, C.; Arnheiter, H. MITF-the first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef]
- Seberg, H.; Van, O.; Cornell, R. Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res. 2017, 30, 454–466. [Google Scholar] [CrossRef]
- Gelmi, M.; Houtzagers, L.; Strub, T.; Krossa, I.; Jager, M. MITF in Normal Melanocytes, Cutaneous and Uveal Melanoma: A Delicate Balance. Int. J. Mol. Sci. 2022, 23, 6001. [Google Scholar] [CrossRef]
- Ferguson, J.; Wilcock, D.; McEntegart, S.; Badrock, A.; Levesque, M.; Dummer, R.; Wellbrock, C.; Smith, M. Osteoblasts contribute to a protective niche that supports melanoma cell proliferation and survival. Pigment Cell Melanoma Res. 2020, 33, 74–85. [Google Scholar] [CrossRef]
- Hauswirth, R.; Haase, B.; Blatter, M.; Brooks, S.; Burger, D.; Drögemüller, C.; Gerber, V.; Henke, D.; Janda, J.; Jude, R.; et al. Mutations in MITF and PAX3 cause “splashed white” and other white spotting phenotypes in horses. PLoS Genet. 2012, 8, e1002653. [Google Scholar] [CrossRef]
- Johnston, S.; McEwan, J.; Pickering, N.; Kijas, J.; Beraldi, D.; Pilkington, J.; Pemberton, J.; Slate, J. Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Mol. Ecol. 2011, 20, 2555–2566. [Google Scholar] [CrossRef]
- Shariflou, M.; Wade, C.; Kijas, J.; McCulloch, R.; Windsor, P.; Tammen, I.; Nicholas, F. Brachygnathia, cardiomegaly and renal hypoplasia syndrome (BCRHS) in Merino sheep maps to a 1.1-megabase region on ovine chromosome OAR2. Anim. Genet. 2013, 44, 231–233. [Google Scholar] [CrossRef]
- Zhou, S.; Zeng, H.; Huang, J.; Lei, L.; Tong, X.; Li, S.; Zhou, Y.; Guo, H.; Khan, M.; Luo, L.; et al. Epigenetic regulation of melanogenesis. Ageing Res. Rev. 2021, 69, 101349. [Google Scholar] [CrossRef]
- Mo, X.; Kazmi, H.; Preston-Alp, S.; Zhou, B.; Zaidi, M. Interferon-gamma induces melanogenesis via post-translational regulation of tyrosinase. Pigment Cell Melanoma Res. 2022, 35, 342–355. [Google Scholar] [CrossRef]
- Ma, S.; Liu, H.; Wang, J.; Wang, L.; Xi, Y.; Liu, Y.; Xu, Q.; Hu, J.; Han, C.; Bai, L.; et al. Transcriptome Analysis Reveals Genes Associated With Sexual Dichromatism of Head Feather Color in Mallard. Front Genet. 2021, 12, 627974. [Google Scholar] [CrossRef]
- Liu, Q.; Qi, Y.; Liang, Q.; Song, J.; Liu, J.; Li, W.; Shu, Y.; Tao, M.; Zhang, C.; Qin, Q.; et al. Targeted disruption of tyrosinase causes melanin reduction in Carassius auratus cuvieri and its hybrid progeny. Sci. China Life Sci. 2019, 62, 1194–1202. [Google Scholar] [CrossRef]
- Van, L.; Nguyen, M.; Braunschweig, M.; Nezer, C.; Collette, C.; Moreau, L.; Archibald, A.; Haley, C.; Buys, N.; Tally, M.; et al. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 2003, 425, 832–836. [Google Scholar]
- Hill, M.; Vande, Z.; Wittkopp, P. Molecular and evolutionary processes generating variation in gene expression. Nat. Rev. Genet. 2021, 22, 203–215. [Google Scholar] [CrossRef]
- Wang, X.; Zhong, J.; Gao, Y.; Ju, Z.; Huang, J. A SNP in intron 8 of CD46 causes a novel transcript associated with mastitis in Holsteins. BMC Genom. 2014, 15, 630. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | Black | Grey | Amber |
|---|---|---|---|
| Ram | 1.236 | 2.85 | 3.165 |
| Ewe | 4.169 | 5.59 | 6.75 |
| Total | 7.405 | 8.144 | 9.240 |
| Sex | Black | Grey | Amber |
|---|---|---|---|
| Ram | 78 | 22 | 33 |
| Ewe | 44 | 16 | 19 |
| Total | 122 | 38 | 52 |
| Sex | Black | Grey | Amber |
|---|---|---|---|
| Ram | 158 | 63 | 132 |
| Ewe | 125 | 43 | 56 |
| Total | 283 | 106 | 186 |
| Variant ID | Primer Sequences (5′ to 3′) | Location |
|---|---|---|
| rs417454260 | F:GGCCACCTGATGTGAAGAAC | Intron 14 |
| R:TTGACAGTGTTGTGGGGCA |
| Genotype | Number of Individuals | Number of Individuals with Different Hoof Colors | ||
|---|---|---|---|---|
| Amber | Grey | Black | ||
| AA | 26 | 19 | 3 | 4 |
| AG | 84 | 21 | 19 | 44 |
| GG | 102 | 12 | 16 | 74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, P.; Wu, H.; Huang, Y.; Lu, X.; Yin, J.; Zhang, Q.; Lan, X. The Hoof Color of Australian White Sheep Is Associated with Genetic Variation of the MITF Gene. Animals 2023, 13, 3218. https://doi.org/10.3390/ani13203218
Su P, Wu H, Huang Y, Lu X, Yin J, Zhang Q, Lan X. The Hoof Color of Australian White Sheep Is Associated with Genetic Variation of the MITF Gene. Animals. 2023; 13(20):3218. https://doi.org/10.3390/ani13203218
Chicago/Turabian StyleSu, Peng, Hui Wu, Yangming Huang, Xiaofang Lu, Jing Yin, Qingfeng Zhang, and Xianyong Lan. 2023. "The Hoof Color of Australian White Sheep Is Associated with Genetic Variation of the MITF Gene" Animals 13, no. 20: 3218. https://doi.org/10.3390/ani13203218