PacBio Full-Length Transcriptome of a Tetraploid Sinocyclocheilus multipunctatus Provides Insights into the Evolution of Cavefish



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
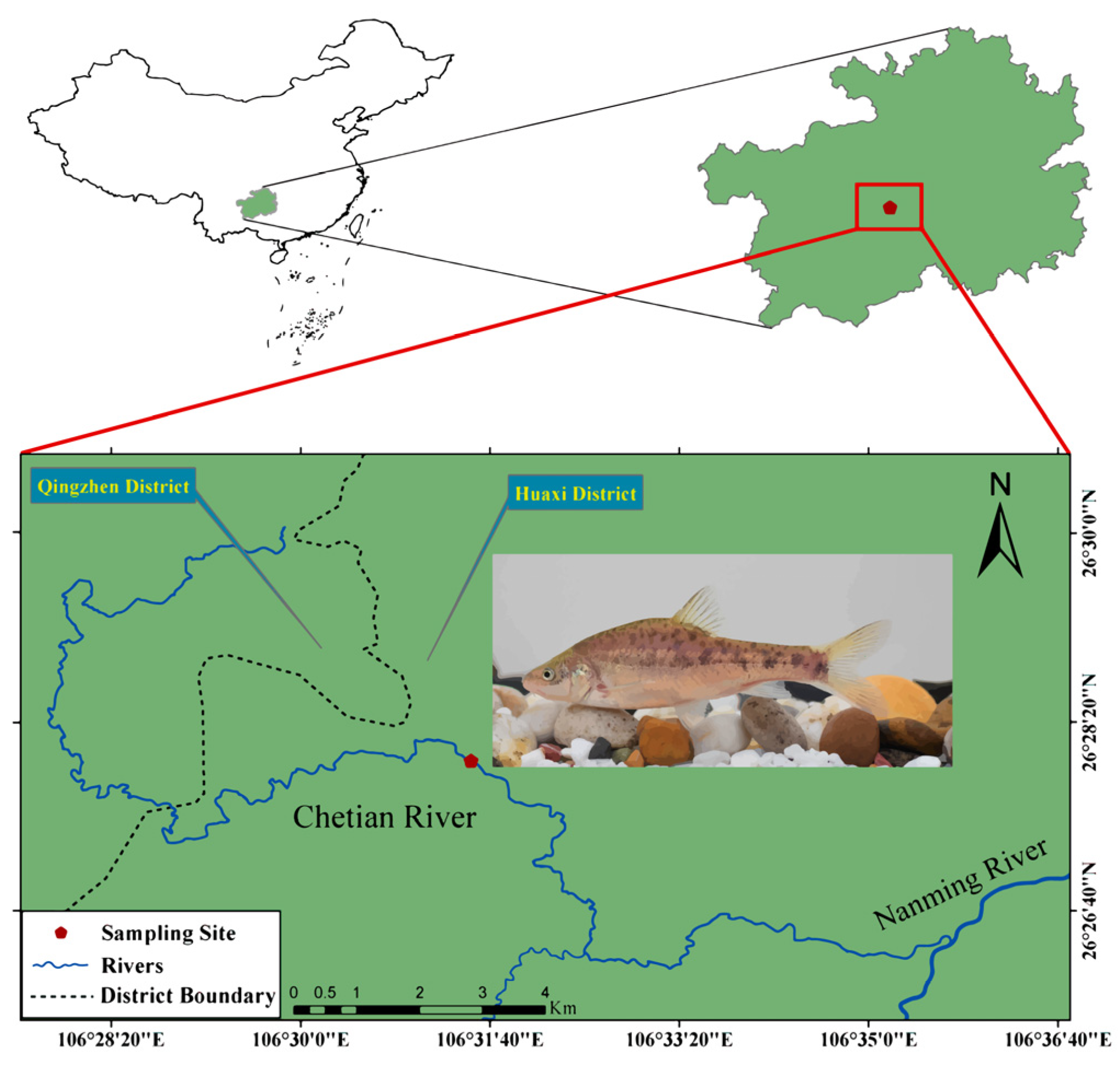
2.1. Sample Collection and RNA Preparation
2.2. PacBio Iso-Seq Library Preparation and Sequencing
2.3. PacBio Iso-Seq Data Processing
2.4. Functional Annotation of Transcripts
2.5. Gene Structure Analysis and Annotation
2.6. Phylogenetic Analysis
2.7. Detection of Polyploidization Events
2.8. Positive Selection Analysis
3. Results
3.1. Summary of FL Reference Transcriptome
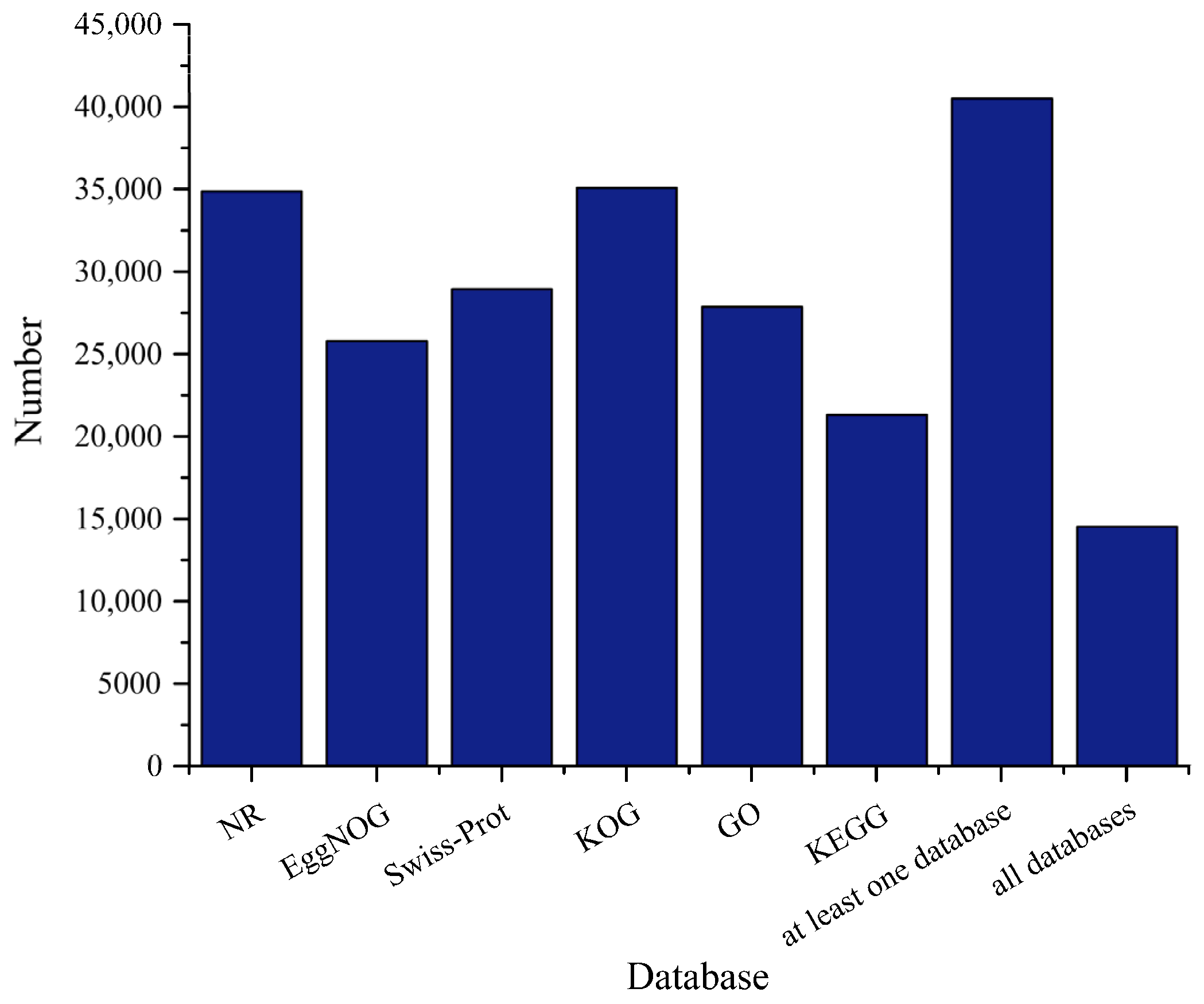
3.2. Basic Annotation of Transcripts
3.3. Detection of SSRs, TFs, and LncRNAs
3.4. Identification of Orthologous Genes and Phylogenetic Tree
3.5. Genome Expansion in S. multipunctatus
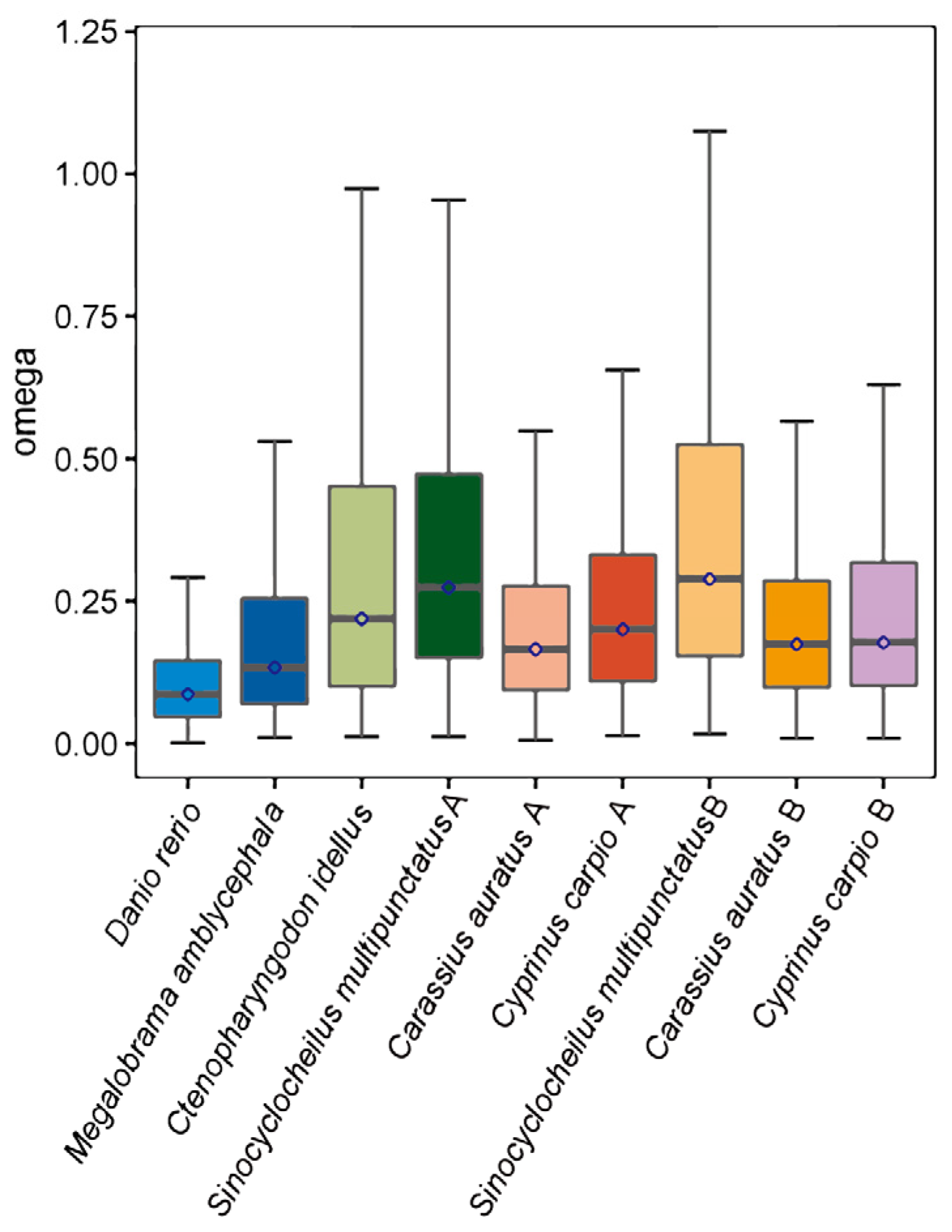
3.6. Accelerated Evolution and Positively Selective Genes in S. multipunctatus
4. Discussion
4.1. Long-Read Reference Reconstruction of the Full-Length Transcripts
4.2. Evolutionary Status and Positive Selection
4.3. The Whole-Genome Duplication Event in S. multipunctatus
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qian, X.; Ba, Y.; Zhuang, Q.; Zhong, G. RNA-Seq Technology and Its Application in Fish Transcriptomics. OMICS J. Integr. Biol. 2014, 18, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, L.; Huang, S.; Wang, G. Full-length transcriptome sequencing of Heliocidaris crassispina using PacBio single-molecule real-time sequencing. Fish Shellfish Immunol. 2022, 120, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Deng, A.; Li, J.; Yao, Z.; Afriyie, G.; Chen, Z.; Guo, Y.; Luo, J.; Wang, Z. SMRT Sequencing of the Full-Length Transcriptome of the Coelomactra antiquata. Front. Genet. 2021, 12, 741243. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Luo, Q.; Tang, Q.; Deng, L.; Zhang, R.; Li, Y. Comprehensive transcripts analysis based on single-molecule real-time sequencing and Illumina sequencing provides insights into the mining of Toll-like receptor family in Schizothorax lissolabiatus. Fish Shellfish Immunol. 2023, 140, 108963. [Google Scholar] [CrossRef]
- Luo, H.; Liu, H.; Zhang, J.; Hu, B.; Zhou, C.; Xiang, M.; Yang, Y.; Zhou, M.; Jing, T.; Li, Z.; et al. Full-length transcript sequencing accelerates the transcriptome research of Gymnocypris namensis, an iconic fish of the Tibetan Plateau. Sci. Rep. 2020, 10, 9668. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, P.; Mo, Q.; Luo, W.; Du, Z.; Jiang, J.; Yang, S.; Zhao, L.; Gong, Q.; Wang, Y. Comprehensive analysis of full-length transcriptomes of Schizothorax prenanti by single-molecule long-read sequencing. Genomics 2022, 114, 456–464. [Google Scholar] [CrossRef]
- Luo, J.; Chai, J.; Wen, Y.; Tao, M.; Lin, G.; Liu, X.; Ren, L.; Chen, Z.; Wu, S.; Li, S. From asymmetrical to balanced genomic diversification during rediploidization: Subgenomic evolution in allotetraploid fish. Sci. Adv. 2020, 6, eaaz7677. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, C. Endemic Fishes of Sinocyclocheilus (Cypriniformes: Cyprinidae) in China-Species Diversity, Cave Adaptation, Systematics and Zoogeography; Science Press: Beijing, China, 2009. [Google Scholar]
- Jeffery, W.R. Cavefish as a Model System in Evolutionary Developmental Biology. Dev. Biol. 2001, 231, 1–12. [Google Scholar] [CrossRef]
- Luo, Q. Phylogeny and Biogeography of Sinocyclocheilus Based on Mitochondrial Genomes; Guizhou Normal University: Guiyang, China, 2023. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, C. Past research and future development on endemic Chinese cavefish of the genus Sinocyclocheilus (Cypriniformes, Cyprinidae). Acta Zootaxonomica Sin. 2006, 31, 769–777. [Google Scholar]
- Wu, Y.; Lü, K. On the systematic status of some Schizothoracin fishes from Guizhou Province, China. Acta Zootaxonomica Sin. 1983, 8, 335–336. [Google Scholar]
- Yang, N.; Li, Y.; Liu, Z.; Chen, Q.; Shen, Y. Molecular phylogenetics and evolutionary history of Sinocyclocheilus (Cypriniformes: Cyprinidae) species within Barbinae in China. Environ. Biol. Fishes 2021, 104, 1149–1162. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, J.; Wang, Y.; Zhang, E.; Zhang, Y.; Li, L.; Xie, F.; Cai, B.; Cao, L.; Zheng, G. Red List of China’s Vertebrates. Biodivers Sci. 2016, 24, 500–551. [Google Scholar] [CrossRef]
- Sahlin, K.; Medvedev, P. De novo clustering of long-read transcriptome data using a greedy, quality value-based algorithm. J. Comput. Biol. 2020, 27, 472–484. [Google Scholar] [CrossRef]
- Gaëta, B.A. BLAST on the Web. Biotechniques 2000, 28, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jia, Y.; Zhu, R.; Chen, K.; Chen, Y. Characterization and analysis of the transcriptome in Gymnocypris selincuoensis on the Qinghai-Tibetan Plateau using single-molecule long-read sequencing and RNA-seq. DNA Res. 2019, 26, 353–363. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, T.; Liu, C.; Song, S.; Zhang, X.; Liu, W.; Jia, H.; Xue, Y.; Guo, A. AnimalTFDB 2.0: A resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2015, 43, D76–D81. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L. GMATA: An Integrated Software Package for Genome-Scale SSR Mining, Marker Development and Viewing. Front. Plant Sci. 2016, 7, 1350. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinf. 2014, 15, 311. [Google Scholar] [CrossRef]
- Kang, Y.; Yang, D.; Kong, L.; Hou, M.; Meng, Y.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Q.; Huang-Yang, M.; Li, Q.; Cui, M.; Dong, Z.; Wang, H.; Yu, J.; Zhao, Y.; Yang, C.; et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat. Genet. 2021, 53, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder2: Fast and accurate phylogenomic orthology analysis from gene sequences. BioRxiv 2019, 20, 238. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Liu, H.; Chen, C.; Gao, Z.; Min, J.; Gu, Y.; Jian, J.; Jiang, X.; Cai, H.; Ebersberger, I.; Xu, M. The draft genome of blunt snout bream (Megalobrama amblycephala) reveals the development of intermuscular bone and adaptation to herbivorous diet. Gigascience 2017, 6, gix039. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-T.; Li, J.-T.; Zhang, X.-F.; Sun, X.-W. Transcriptome analysis reveals the time of the fourth round of genome duplication in common carp (Cyprinus carpio). BMC Genom. 2012, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Ma, Z.-Y.; Zheng, G.-D.; Zou, S.-M.; Zhang, X.-J.; Zhang, Y.-A. Chromosome-level genome assembly of grass carp (Ctenopharyngodon idella) provides insights into its genome evolution. BMC Genom. 2022, 23, 271. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, X.; Wang, X.; Li, J.; Liu, G.; Kuang, Y.; Xu, J.; Zheng, X.; Ren, L.; Wang, G. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Y.; Chen, Q.; Sun, Y.; Lu, Z. WGDdetector: A pipeline for detecting whole genome duplication events using the genome or transcriptome annotations. BMC Bioinf. 2019, 20, 75. [Google Scholar] [CrossRef]
- Pardos-Blas, J.R.; Irisarri, I.; Abalde, S.; Afonso, C.M.; Tenorio, M.J.; Zardoya, R. The genome of the venomous snail Lautoconus ventricosus sheds light on the origin of conotoxin diversity. Gigascience 2021, 10, giab037. [Google Scholar] [CrossRef]
- Gaikwad, K.; Ramakrishna, G.; Srivastava, H.; Saxena, S.; Kaila, T.; Tyagi, A.; Sharma, P.; Sharma, S.; Sharma, R.; Mahla, H. The chromosome-scale genome assembly of cluster bean provides molecular insight into edible gum (galactomannan) biosynthesis family genes. Sci. Rep. 2023, 13, 9941. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Wen, Y.; Chai, J.; Ma, W.; Murphy, R.W.; He, S.; Chen, Z.; Zhang, Y.; Lu, X. Polyploidization, hybridization, and maternal and paternal lineages in Cyprinids (Teleostei: Cypriniformes). Res. Sq. 2020, 4, 1–21. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, Y.; Zhang, Y.; Ning, Z.; Li, Y.; Zhao, Q.; Lu, H.; Huang, R.; Xia, X.; Feng, Q.; et al. The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nat. Genet. 2015, 47, 625–631. [Google Scholar] [CrossRef]
- Xu, B.; Yang, Z. PAMLX: A graphical user interface for PAML. Mol. Biol. Evol. 2013, 30, 2723–2724. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Y.; Zhang, Z.; He, S. Comprehensive transcriptome analysis reveals accelerated genic evolution in a Tibet fish, Gymnodiptychus pachycheilus. Genome Biol. Evol. 2015, 7, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Ma, X.; He, S. Evidence of high-altitude adaptation in the glyptosternoid fish, Creteuchiloglanis macropterus from the Nujiang River obtained through transcriptome analysis. BMC Evol. Biol. 2017, 17, 229. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.B.-T.; Ulitsky, I. The functions of long noncoding RNAs in development and stem cells. Development 2016, 143, 3882–3894. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Xiao, B.; Guo, J. Acting mechanisms and research methods of long noncoding RNAs. Hereditas 2013, 35, 269–280. [Google Scholar] [CrossRef]
- Fulton, D.L.; Sundararajan, S.; Badis, G.; Hughes, T.R.; Wasserman, W.W.; Roach, J.C.; Sladek, R. TFCat: The curated catalog of mouse and human transcription factors. Genome Biol. 2009, 10, R29. [Google Scholar] [CrossRef]
- Thomas, J.H.; Emerson, R.O. Evolution of C2H2-zinc finger genes revisited. BMC Evol. Biol. 2009, 9, 51. [Google Scholar] [CrossRef]
- O’Connell, M.; Wright, J.M. Microsatellite DNA in fishes. Rev. Fish Biol. Fish. 1997, 7, 331–363. [Google Scholar] [CrossRef]
- Han, Z.; Xiao, S.; Li, W.; Ye, K.; Wang, Z.Y. The identification of growth, immune related genes and marker discovery through transcriptome in the yellow drum (Nibea albiflora). Genes Genom. 2018, 40, 881–891. [Google Scholar] [CrossRef]
- Li, C.; Teng, T.; Shen, F.; Guo, J.; Chen, Y.; Zhu, C.; Ling, Q. Transcriptome characterization and SSR discovery in Squaliobarbus curriculus. J. Oceanol. Limnol. 2019, 37, 235–244. [Google Scholar] [CrossRef]
- Luo, W.; Cao, X.; Xu, X.; Huang, S.; Liu, C.; Tomljanovic, T. Developmental transcriptome analysis and identification of genes involved in formation of intestinal air-breathing function of Dojo loach, Misgurnus anguillicaudatus. Sci. Rep. 2016, 6, 31845. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, L.; Chen, S.; Zan, R.; Xiao, H.; Zhang, Y.-p. The complete mitochondrial genomes of two species from Sinocyclocheilus (Cypriniformes: Cyprinidae) and a phylogenetic analysis within Cyprininae. Mol. Biol. Rep. 2010, 37, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Kosiol, C.; Vinar, T.; da Fonseca, R.R.; Hubisz, M.J.; Bustamante, C.D.; Nielsen, R.; Siepel, A. Patterns of positive selection in six Mammalian genomes. PLoS Genet. 2008, 4, e1000144. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Zhou, X.; Li, J.; Zhang, M.; Luo, S. Full-length transcriptome of Misgurnus anguillicaudatus provides insights into evolution of genus Misgurnus. Sci. Rep. 2018, 8, 11699. [Google Scholar] [CrossRef]
- Stanley, S.M. A theory of evolution above the species level. Proc. Natl. Acad. Sci. USA 1975, 72, 646–650. [Google Scholar] [CrossRef]
- Sato, Y.; Nishida, M. Teleost fish with specific genome duplication as unique models of vertebrate evolution. Environ. Biol. Fishes 2010, 88, 169–188. [Google Scholar] [CrossRef]
- Xiao, S.; Mou, Z.; Fan, D.; Zhou, H.; Zou, M.; Zou, Y.; Zhou, C.; Yang, R.; Liu, J.; Zhu, S.; et al. Genome of Tetraploid Fish Schizothorax o’connori Provides Insights into Early Re-diploidization and High-Altitude Adaptation. iScience 2020, 23, 101497. [Google Scholar] [CrossRef]
- Yin, A.; Harrison, T.M. Geologic Evolution of the Himalayan-Tibetan Orogen. Annu. Rev. Earth Planet. Sci. 2000, 28, 211–280. [Google Scholar] [CrossRef]
- David, L.; Blum, S.; Feldman, M.W.; Lavi, U.; Hillel, J. Recent Duplication of the Common Carp (Cyprinus carpio L.) Genome as Revealed by Analyses of Microsatellite Loci. Mol. Biol. Evol. 2003, 20, 1425–1434. [Google Scholar] [CrossRef]
- Larhammar, D.; Risinger, C. Molecular Genetic Aspects of Tetraploidy in the Common Carp Cyprinus carpio. Mol. Phylogenetics Evol. 1994, 3, 59–68. [Google Scholar] [CrossRef]
- Xu, P.; Xu, J.; Liu, G.; Chen, L.; Zhou, Z.; Peng, W.; Jiang, Y.; Zhao, Z.; Jia, Z.; Sun, Y.; et al. The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat. Commun. 2019, 10, 4625. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Items | Number |
|---|---|---|
| Subreads | Subreads base (G) | 259 |
| Number of subreads | 76,978,307 | |
| Average length (bp) | 1820 | |
| N50 length (bp) | 1957 | |
| CCS reads | Number of reads | 1,776,276 |
| Average length (bp) | 1950 | |
| N50 length (bp) | 2081 | |
| FLNC reads | Number of reads | 1,554,240 |
| Average length (bp) | 1827 | |
| N50 length (bp) | 1959 | |
| Full-length transcriptome | transcripts number | 232,126 |
| Average length (bp) | 2075 | |
| N50 length (bp) | 2338 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Duan, Q.; Luo, Q.; Deng, L. PacBio Full-Length Transcriptome of a Tetraploid Sinocyclocheilus multipunctatus Provides Insights into the Evolution of Cavefish. Animals 2023, 13, 3399. https://doi.org/10.3390/ani13213399
Zhang R, Duan Q, Luo Q, Deng L. PacBio Full-Length Transcriptome of a Tetraploid Sinocyclocheilus multipunctatus Provides Insights into the Evolution of Cavefish. Animals. 2023; 13(21):3399. https://doi.org/10.3390/ani13213399
Chicago/Turabian StyleZhang, Renyi, Qian Duan, Qi Luo, and Lei Deng. 2023. "PacBio Full-Length Transcriptome of a Tetraploid Sinocyclocheilus multipunctatus Provides Insights into the Evolution of Cavefish" Animals 13, no. 21: 3399. https://doi.org/10.3390/ani13213399
APA StyleZhang, R., Duan, Q., Luo, Q., & Deng, L. (2023). PacBio Full-Length Transcriptome of a Tetraploid Sinocyclocheilus multipunctatus Provides Insights into the Evolution of Cavefish. Animals, 13(21), 3399. https://doi.org/10.3390/ani13213399