
Genomic Selection for Live Weight in the 14th Month in Alpine Merino Sheep Combining GWAS Information
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotype and Genotype Data
2.2. GWAS Model Analysis Methods
2.3. GS Model Analysis Method
2.4. Calculation Method of GS Accuracy
3. Results
3.1. Statistics and Distribution of Phenotypic Data
3.2. Call Rate and Distribution of Genotype Data
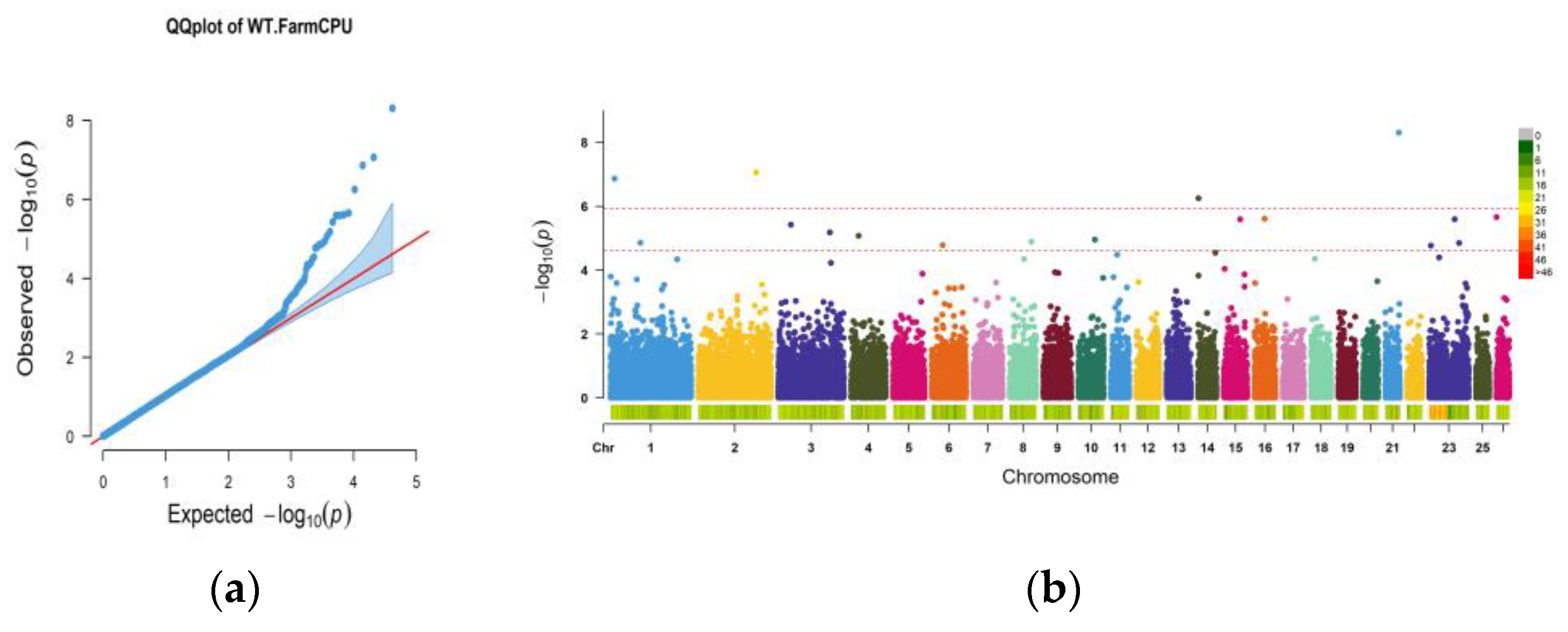
3.3. GWAS Analysis Results
3.4. GS Analysis Results
4. Discussion
4.1. Animals and Data
4.2. GWAS and Candidate Genes
4.3. Comparative Analysis of the Accuracy of Different Models of Genome Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Visscher, P.M.; Goddard, M.E. Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. 2009, 91, 47–60. [Google Scholar] [CrossRef]
- Habier, D.; Tetens, J.; Seefried, F.R.; Lichtner, P.; Thaller, G. The impact of genetic relationship information on genomic breeding values in German Holstein cattle. Genet. Sel. Evol. 2010, 42, 5. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, L.R. Strategy for applying genome-wide selection in dairy cattle. J. Anim. Breed. Genet. 2006, 123, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.E.; Hayes, B.J. Genomic selection. J. Anim. Breed. Genet. 2007, 124, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Esfandyari, H.; Sørensen, A.C.; Bijma, P. A crossbred reference population can improve the response to genomic selection for crossbred performance. Genet. Sel. Evol. 2015, 47, 76. [Google Scholar] [CrossRef]
- Song, H.; Ye, S.; Jiang, Y.; Zhang, Z.; Zhang, Q.; Ding, X. Using imputation-based whole-genome sequencing data to improve the accuracy of genomic prediction for combined populations in pigs. Genet. Sel. Evol. 2019, 51, 58. [Google Scholar] [CrossRef]
- Lourenco, D.A.; Tsuruta, S.; Fragomeni, B.O.; Masuda, Y.; Aguilar, I.; Legarra, A.; Bertrand, J.K.; Amen, T.S.; Wang, L.; Moser, D.W.; et al. Genetic evaluation using single-step genomic best linear unbiased predictor in American Angus. J. Anim. Sci. 2015, 93, 2653–2662. [Google Scholar] [CrossRef]
- Zhao, Z.D.; Zhang, L. Applications of genome selection in sheep breeding. Yi Chuan 2019, 41, 293–303. [Google Scholar]
- Wolc, A.; Zhao, H.H.; Arango, J.; Settar, P.; Fulton, J.E.; O’Sullivan, N.P.; Preisinger, R.; Stricker, C.; Habier, D.; Fernando, R.L.; et al. Response and inbreeding from a genomic selection experiment in layer chickens. Genet. Sel. Evol. 2015, 47, 59. [Google Scholar] [CrossRef]
- Lopes, M.S.; Bovenhuis, H.; van Son, M.; Nordbø, Ø.; Grindflek, E.H.; Knol, E.F.; Bastiaansen, J.W. Using markers with large effect in genetic and genomic predictions. J. Anim. Sci. 2017, 95, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Liu, C. Encyclopedia of Modern Sheep Production Technology; Agriculture Press: Beijing, China, 2014. [Google Scholar]
- Zhang, L.; Liu, J.; Zhao, F.; Ren, H.; Xu, L.; Lu, J.; Zhang, S.; Zhang, X.; Wei, C.; Lu, G.; et al. Genome-wide association studies for growth and meat production traits in sheep. PLoS ONE 2013, 8, e66569. [Google Scholar] [CrossRef] [PubMed]
- Kominakis, A.; Hager-Theodorides, A.L.; Zoidis, E.; Saridaki, A.; Antonakos, G.; Tsiamis, G. Combined GWAS and ‘guilt by association’-based prioritization analysis identifies functional candidate genes for body size in sheep. Genet. Sel. Evol. 2017, 49, 41. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, A.A.; Deniskova, T.E.; Yudin, N.S.; Dotsev, A.V.; Khamiruev, T.N.; Selionova, M.I.; Egorov, S.V.; Reyer, H.; Wimmers, K.; Brem, G.; et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genom. 2019, 20, 294. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Zamani, P.; Vatankhah, M.; Abdoli, R. Genome-wide association study of birth weight in sheep. Animal 2019, 13, 1797–1803. [Google Scholar] [CrossRef]
- de Los Campos, G.; Vazquez, A.I.; Fernando, R.; Klimentidis, Y.C.; Sorensen, D. Prediction of complex human traits using the genomic best linear unbiased predictor. PLoS Genet. 2013, 9, e1003608. [Google Scholar] [CrossRef]
- Zhang, Z.; Ober, U.; Erbe, M.; Zhang, H.; Gao, N.; He, J.; Li, J.; Simianer, H. Improving the accuracy of whole genome prediction for complex traits using the results of genome wide association studies. PLoS ONE 2014, 9, e93017. [Google Scholar] [CrossRef]
- Gao, N.; Li, J.; He, J.; Xiao, G.; Luo, Y.; Zhang, H.; Chen, Z.; Zhang, Z. Improving accuracy of genomic prediction by genetic architecture based priors in a Bayesian model. BMC Genet. 2015, 16, 120. [Google Scholar] [CrossRef]
- Moghaddar, N.; Khansefid, M.; van der Werf, J.H.J.; Bolormaa, S.; Duijvesteijn, N.; Clark, S.A.; Swan, A.A.; Daetwyler, H.D.; MacLeod, I.M. Genomic prediction based on selected variants from imputed whole-genome sequence data in Australian sheep populations. Genet. Sel. Evol. 2019, 51, 72. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Bland, J.M.; Altman, D.G. Multiple significance tests: The Bonferroni method. BMJ 1995, 310, 170. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy. Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.; Cullis, B.; Gilmour, A.; Gogel, B.; Thompson, R. ASReml-R Reference Manual Version 4; VSN International Ltd.: Hemel Hempstead, UK, 2017. [Google Scholar]
- Browne, M.W. Cross-Validation Methods. J. Math. Psychol. 2000, 44, 108–132. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Choi, T.J.; Kim, Y.S.; Lee, Y.M.; Zahangir Alam, M.; Jung, J.H.; Choe, H.S.; Kim, J.J. Comparison of genomic predictions for carcass and reproduction traits in Berkshire, Duroc and Yorkshire populations in Korea. Asian-Australas. J. Anim. Sci. 2019, 32, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Bolormaa, S.; Gore, K.; van der Werf, J.H.; Hayes, B.J.; Daetwyler, H.D. Design of a low-density SNP chip for the main Australian sheep breeds and its effect on imputation and genomic prediction accuracy. Anim. Genet. 2015, 46, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Naserkheil, M.; Lee, D.H.; Mehrban, H. Improving the accuracy of genomic evaluation for linear body measurement traits using single-step genomic best linear unbiased prediction in Hanwoo beef cattle. BMC Genet. 2020, 21, 144. [Google Scholar] [CrossRef]
- Cao, Y.; Song, X.; Shan, H.; Jiang, J.; Xiong, P.; Wu, J.; Shi, F.; Jiang, Y. Genome-Wide Association Study of Body Weights in Hu Sheep and Population Verification of Related Single-Nucleotide Polymorphisms. Front. Genet. 2020, 11, 588. [Google Scholar] [CrossRef]
- Tuersuntuoheti, M.; Zhang, J.; Zhou, W.; Zhang, C.L.; Liu, C.; Chang, Q.; Liu, S. Exploring the growth trait molecular markers in two sheep breeds based on Genome-wide association analysis. PLoS ONE 2023, 18, e0283383. [Google Scholar] [CrossRef]
- Moaeen-Ud-Din, M.; Danish Muner, R.; Khan, M.S. Genome wide association study identifies novel candidate genes for growth and body conformation traits in goats. Sci. Rep. 2022, 12, 9891. [Google Scholar] [CrossRef]
- Easa, A.A.; Selionova, M.; Aibazov, M.; Mamontova, T.; Sermyagin, A.; Belous, A.; Abdelmanova, A.; Deniskova, T.; Zinovieva, N. Identification of Genomic Regions and Candidate Genes Associated with Body Weight and Body Conformation Traits in Karachai Goats. Genes 2022, 13, 1773. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, F.; Gao, G.; Yan, X.; Liu, H.; Liu, Z.; Wang, Z.; He, L.; Lv, Q.; Wang, Z.; et al. Genome-Wide Association Study of Body Weight Traits in Inner Mongolia Cashmere Goats. Front. Vet. Sci. 2021, 8, 752746. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bai, C.; Shi, L.; He, Y.; Hu, M.; Sun, H.; Peng, H.; Lai, W.; Jiao, S.; Zhao, Z.; et al. Detection of selection signatures in South African Mutton Merino sheep using whole-genome sequencing data. Anim. Genet. 2022, 53, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, H.; Raffat, A.; Moradi, H.; Shoja, J.; Moradi, M.H. Estimation of linkage disequilibrium and whole-genome scan for detection of loci under selection associated with body weight in Zandi sheep breed. Agric. Biotechnol. J. 2018, 9, 151–172. [Google Scholar]
- Liu, Y.; Li, H.; Wang, M.; Zhang, X.; Yang, L.; Zhao, C.; Wu, C. Genetic architectures and selection signatures of body height in Chinese indigenous donkeys revealed by next-generation sequencing. Anim. Genet. 2022, 53, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.X.; Fan, Q.C.; Wang, J.Y.; Zhang, T.; Xue, Q.; Shi, H.Q. Genome-wide association study on reproductive traits in Jinghai Yellow Chicken. Anim. Reprod. Sci. 2015, 163, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.F.; Chen, Y.J.; Yang, Z.Q.; Shi, K.; Chen, C.K. A genome-wide association study of growth trait-related single nucleotide polymorphisms in Chinese Yancheng chickens. Genet. Mol. Res. 2015, 14, 15783–15792. [Google Scholar] [CrossRef]
- Smith, J.L.; Wilson, M.L.; Nilson, S.M.; Rowan, T.N.; Schnabel, R.D.; Decker, J.E.; Seabury, C.M. Genome-wide association and genotype by environment interactions for growth traits in U.S. Red Angus cattle. BMC Genom. 2022, 23, 517. [Google Scholar] [CrossRef]
- Posbergh, C.J.; Huson, H.J. All sheeps and sizes: A genetic investigation of mature body size across sheep breeds reveals a polygenic nature. Anim. Genet. 2021, 52, 99–107. [Google Scholar] [CrossRef]
- Al-Mamun, H.A.; Kwan, P.; Clark, S.A.; Ferdosi, M.H.; Tellam, R.; Gondro, C. Genome-wide association study of body weight in Australian Merino sheep reveals an orthologous region on OAR6 to human and bovine genomic regions affecting height and weight. Genet. Sel. Evol. 2015, 47, 66. [Google Scholar] [CrossRef]
- Xia, J.; Fan, H.; Chang, T.; Xu, L.; Zhang, W.; Song, Y.; Zhu, B.; Zhang, L.; Gao, X.; Chen, Y.; et al. Searching for new loci and candidate genes for economically important traits through gene-based association analysis of Simmental cattle. Sci. Rep. 2017, 7, 42048. [Google Scholar] [CrossRef]
- Liu, Y.; Duan, X.; Chen, S.; He, H.; Liu, X. NCAPG is differentially expressed during longissimus muscle development and is associated with growth traits in Chinese Qinchuan beef cattle. Genet. Mol. Biol. 2015, 38, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Lindholm-Perry, A.K.; Sexten, A.K.; Kuehn, L.A.; Smith, T.P.; King, D.A.; Shackelford, S.D.; Wheeler, T.L.; Ferrell, C.L.; Jenkins, T.G.; Snelling, W.M.; et al. Association, effects and validation of polymorphisms within the NCAPG-LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Staiger, E.A.; Al Abri, M.A.; Pflug, K.M.; Kalla, S.E.; Ainsworth, D.M.; Miller, D.; Raudsepp, T.; Sutter, N.B.; Brooks, S.A. Skeletal variation in Tennessee Walking Horses maps to the LCORL/NCAPG gene region. Physiol. Genom. 2016, 48, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Tetens, J.; Widmann, P.; Kühn, C.; Thaller, G. A genome-wide association study indicates LCORL/NCAPG as a candidate locus for withers height in German Warmblood horses. Anim. Genet. 2013, 44, 467–471. [Google Scholar] [CrossRef]
- Hoshiba, H.; Setoguchi, K.; Watanabe, T.; Kinoshita, A.; Mizoshita, K.; Sugimoto, Y.; Takasuga, A. Comparison of the effects explained by variations in the bovine PLAG1 and NCAPG genes on daily body weight gain, linear skeletal measurements and carcass traits in Japanese Black steers from a progeny testing program. Anim. Sci. J. 2013, 84, 529–534. [Google Scholar] [CrossRef]
- Naserkheil, M.; Manzari, Z.; Dang, C.G.; Lee, S.S.; Park, M.N. Exploring and Identifying Candidate Genes and Genomic Regions Related to Economically Important Traits in Hanwoo Cattle. Curr. Issues Mol. Biol. 2022, 44, 6075–6092. [Google Scholar] [CrossRef]
- Matika, O.; Riggio, V.; Anselme-Moizan, M.; Law, A.S.; Pong-Wong, R.; Archibald, A.L.; Bishop, S.C. Genome-wide association reveals QTL for growth, bone and in vivo carcass traits as assessed by computed tomography in Scottish Blackface lambs. Genet. Sel. Evol. 2016, 48, 11. [Google Scholar] [CrossRef]
- He, S.; Di, J.; Han, B.; Chen, L.; Liu, M.; Li, W. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes Related to Economically Important Traits in Chinese Merino. Animals 2020, 10, 524. [Google Scholar] [CrossRef]
- Hu, L.; Xiao, Y.; Xiong, Z.; Zhao, F.; Yin, C.; Zhang, Y.; Su, P.; Li, D.; Chen, Z.; Ma, X.; et al. MACF1, versatility in tissue-specific function and in human disease. Semin. Cell Dev. Biol. 2017, 69, 3–8. [Google Scholar] [CrossRef]
- Jiayuan, M.; Yujie, L.; Kuirong, C.; Siran, Z.; Wenjing, Q.; Lingli, F.; Xiaoxiao, L.; Liang, L.; Ganqiu, L.; Jing, L. Identifying selection signatures and runs of homozygosity for spine curvature in Chinese indigenous pigs. Anim. Genet. 2022, 53, 513–517. [Google Scholar] [CrossRef]
- Selionova, M.; Aibazov, M.; Mamontova, T.; Malorodov, V.; Sermyagin, A.; Zinovyeva, N.; Easa, A.A. Genome-wide association study of live body weight and body conformation traits in young Karachai goats. Small Rumin. Res. 2022, 216, 106836. [Google Scholar] [CrossRef]
- Xiao, P.; Goodarzi, P.; Pezeshki, A.; Hagen, D.E. RNA-seq reveals insights into molecular mechanisms of metabolic restoration via tryptophan supplementation in low birth weight piglet model. J. Anim. Sci. 2022, 100, 106836. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.J.; Li, X.H.; Li, G.G.; Hu, Y.; Chen, X.D.; Deng, H.W. Identification of novel pleiotropic gene for bone mineral density and lean mass using the cFDR method. Ann. Hum. Genet. 2021, 85, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Guo, Y.; Zhang, L.; Xu, L.; Gao, X.; Zhu, B.; Gao, H.; Ni, H.; Chen, Y. Multi-strategy genome-wide association studies identify the DCAF16-NCAPG region as a susceptibility locus for average daily gain in cattle. Sci. Rep. 2016, 6, 38073. [Google Scholar] [CrossRef]
- van den Berg, I.; Hayes, B.J.; Chamberlain, A.J.; Goddard, M.E. Overlap between eQTL and QTL associated with production traits and fertility in dairy cattle. BMC Genom. 2019, 20, 291. [Google Scholar] [CrossRef]
- Carson, A.R.; Scherer, S.W. Identifying concerted evolution and gene conversion in mammalian gene pairs lasting over 100 million years. BMC Evol. Biol. 2009, 9, 156. [Google Scholar] [CrossRef]
- Hanson, J.; Brezavar, D.; Hughes, S.; Amudhavalli, S.; Fleming, E.; Zhou, D.; Alaimo, J.T.; Bonnen, P.E. TAB2 variants cause cardiovascular heart disease, connective tissue disorder, and developmental delay. Clin. Genet. 2022, 101, 214–220. [Google Scholar] [CrossRef]
- Lopez, P.H.; Aja, S.; Aoki, K.; Seldin, M.M.; Lei, X.; Ronnett, G.V.; Wong, G.W.; Schnaar, R.L. Mice lacking sialyltransferase ST3Gal-II develop late-onset obesity and insulin resistance. Glycobiology 2017, 27, 129–139. [Google Scholar] [CrossRef]
- Perez, É.S.; Cury, S.S.; Zanella, B.T.T.; Carvalho, R.F.; Duran, B.O.S.; Dal-Pai-Silva, M. Identification of Novel Genes Associated with Fish Skeletal Muscle Adaptation during Fasting and Refeeding Based on a Meta-Analysis. Genes 2022, 13, 2378. [Google Scholar] [CrossRef]
- Zhang, D.; Ran, J.; Li, J.; Yu, C.; Cui, Z.; Amevor, F.K.; Wang, Y.; Jiang, X.; Qiu, M.; Du, H.; et al. miR-21-5p Regulates the Proliferation and Differentiation of Skeletal Muscle Satellite Cells by Targeting KLF3 in Chicken. Genes 2021, 12, 814. [Google Scholar] [CrossRef]
- Edwards, S.M.; Sørensen, I.F.; Sarup, P.; Mackay, T.F.; Sørensen, P. Genomic Prediction for Quantitative Traits Is Improved by Mapping Variants to Gene Ontology Categories in Drosophila melanogaster. Genetics 2016, 203, 1871–1883. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, I.M.; Bowman, P.J.; Vander Jagt, C.J.; Haile-Mariam, M.; Kemper, K.E.; Chamberlain, A.J.; Schrooten, C.; Hayes, B.J.; Goddard, M.E. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, J.; Ding, X.; Bijma, P.; de Koning, D.J.; Zhang, Q. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS ONE 2010, 5, e12648. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Item | Number | Mean | Sd | Median | Trimmed | Mad | Min | Max | Se |
|---|---|---|---|---|---|---|---|---|---|
| All | 1310 | 44.79 | 4.35 | 44.4 | 44.66 | 4.15 | 32.6 | 58.6 | 0.12 |
| Group 1 | 655 | 44.72 | 4.38 | 44.4 | 44.63 | 4.45 | 33.4 | 57.8 | 0.17 |
| Group 2 | 655 | 44.86 | 4.31 | 44.4 | 44.69 | 4.15 | 32.6 | 58.6 | 0.17 |
| Trait | Chr | SNP Name | Position | p-Value | Gene Name | Gene Position |
|---|---|---|---|---|---|---|
| WT | 1 | 1_13705982 | 13,705,982 | 1.38 × 10−7 | MACF1 | 13,541,086–13,881,011 |
| 1 | 1_101812709 | 101,812,709 | 1.39 × 10−5 | SPRR4 | 101,734,532–101,736,907 | |
| 2 | 2_198504883 | 198,504,883 | 8.74 × 10−8 | ANKRD44 | 198,238,519–198,479,012 | |
| 3 | 3_42773188 | 42,773,188 | 3.76 × 10−6 | ACTR2 | 42,872,405–42,911,004 | |
| 3 | 3_42773188 | 42,773,188 | 3.76 × 10−6 | SPRED2 | 42,708,519–42,827,598 | |
| 3 | 3_176412255 | 176,412,255 | 6.67 × 10−6 | TRNAC | 176,470,024–176,470,095 | |
| 3 | 3_176412255 | 176,412,255 | 6.67 × 10−6 | SYN3 | 175,963,647–176,440,791 | |
| 6 | 6_37211546 | 37,211,546 | 1.65 × 10−5 | DCAF16 | 37,199,219–37,211,835 | |
| 6 | 6_37211546 | 37,211,546 | 1.65 × 10−5 | FAM184B | 37,061,089–37,179,847 | |
| 6 | 6_37211546 | 37,211,546 | 1.65 × 10−5 | NCAPG | 37,179,188–37,257,373 | |
| 6 | 6_37211546 | 37,211,546 | 1.65 × 10−5 | LCORL | 37,274,935–37,426,290 | |
| 8 | 8_73354847 | 73,354,847 | 1.29 × 10−5 | TAB2 | 73,362,589–73,448,254 | |
| 14 | 14_1078888 | 1,078,888 | 5.70 × 10−7 | SF3B3 | 957,834–996,615 | |
| 14 | 14_1078888 | 1,078,888 | 5.70 × 10−7 | COG4 | 996,835–1,022,199 | |
| 14 | 14_1078888 | 1,078,888 | 5.70 × 10−7 | FUK | 1,022,451–1,036,935 | |
| 14 | 14_1078888 | 1,078,888 | 5.70 × 10−7 | ST3GAL2 | 1,052,461–1,101,237 | |
| 14 | 14_1078888 | 1,078,888 | 5.70 × 10−7 | DDX19A | 1,110,280–1,128,800 | |
| 14 | 14_1078888 | 1,078,888 | 5.70 × 10−7 | AARS | 1,165,135–1,185,981 | |
| 15 | 15_56544782 | 56,544,782 | 2.56 × 10−6 | BDNF | 56,424,495–56,488,905 | |
| 16 | 16_35207312 | 35,207,312 | 2.49 × 10−6 | FYB | 35,105,425–35,282,410 | |
| 21 | 21_45980537 | 45,980,537 | 2.96 × 10−9 | CCND1 | 46,064,385–46,074,757 | |
| 26 | 26_499128 | 499,128 | 2.22 × 10−6 | DLGAP2 | 279,898–905,775 |
| Prior Marker Information | Matrix | Genetic Variance | Environmental Variance | Heritability | Weight | Prediction Accuracy | Promotion |
|---|---|---|---|---|---|---|---|
| - | G | 5.427 | 10.751 | 0.335 | - | 0.154 (0.03) | - |
| Top 5% | G1 | 2.702 | 13.308 | 0.169 | 0.333 | - | - |
| G2 | 5.418 | 10.764 | 0.335 | 0.667 | - | - | |
| G3 | 4.840 | 11.229 | 0.301 | - | 0.158 (0.03) | +2.59% | |
| Top 10% | G1 | 4.035 | 12.013 | 0.251 | 0.434 | - | - |
| G2 | 5.256 | 10.915 | 0.325 | 0.566 | - | - | |
| G3 | 5.331 | 10.767 | 0.331 | - | 0.165 (0.02) | +7.14% | |
| Top 15% | G1 | 4.052 | 11.992 | 0.253 | 0.436 | - | - |
| G2 | 5.236 | 10.933 | 0.324 | 0.564 | - | - | |
| G3 | 5.301 | 10.800 | 0.329 | - | 0.164 (0.03) | +6.49% | |
| Top 20% | G1 | 4.786 | 11.319 | 0.297 | 0.483 | - | - |
| G2 | 5.129 | 11.033 | 0.317 | 0.517 | - | - | |
| G3 | 5.493 | 10.626 | 0.341 | - | 0.166 (0.03) | +7.79% |
| Prior Marker Information | Matrix | Genetic Variance | Environmental Variance | Heritability | Weight | Prediction Accuracy | Promotion |
|---|---|---|---|---|---|---|---|
| - | G | 6.176 | 10.351 | 0.374 | - | 0.190 (0.02) | - |
| Top 5% | G1 | 4.463 | 11.960 | 0.272 | 0.428 | - | - |
| G2 | 5.968 | 10.542 | 0.361 | 0.572 | - | - | |
| G3 | 6.296 | 10.174 | 0.382 | - | 0.201 (0.02) | +5.79% | |
| Top 10% | G1 | 4.688 | 11.740 | 0.285 | 0.438 | - | - |
| G2 | 6.012 | 10.503 | 0.364 | 0.562 | - | - | |
| G3 | 6.057 | 10.340 | 0.368 | - | 0.188 (0.02) | −1.05% | |
| Top 15% | G1 | 4.969 | 11.475 | 0.302 | 0.452 | - | - |
| G2 | 6.027 | 10.489 | 0.365 | 0.548 | - | - | |
| G3 | 6.060 | 10.396 | 0.368 | - | 0.184 (0.02) | −3.15% | |
| Top 20% | G1 | 5.288 | 11.194 | 0.321 | 0.469 | - | - |
| G2 | 5.986 | 10.523 | 0.363 | 0.531 | - | - | |
| G3 | 6.139 | 10.330 | 0.373 | - | 0.185 (0.02) | −2.63% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Li, J.; Wang, H.; Zhang, R.; An, X.; Yuan, C.; Guo, T.; Yue, Y. Genomic Selection for Live Weight in the 14th Month in Alpine Merino Sheep Combining GWAS Information. Animals 2023, 13, 3516. https://doi.org/10.3390/ani13223516
Li C, Li J, Wang H, Zhang R, An X, Yuan C, Guo T, Yue Y. Genomic Selection for Live Weight in the 14th Month in Alpine Merino Sheep Combining GWAS Information. Animals. 2023; 13(22):3516. https://doi.org/10.3390/ani13223516
Chicago/Turabian StyleLi, Chenglan, Jianye Li, Haifeng Wang, Rui Zhang, Xuejiao An, Chao Yuan, Tingting Guo, and Yaojing Yue. 2023. "Genomic Selection for Live Weight in the 14th Month in Alpine Merino Sheep Combining GWAS Information" Animals 13, no. 22: 3516. https://doi.org/10.3390/ani13223516