


Boar Seminal Microbiota in Relation to Sperm Quality under Tropical Environments

 , , , , , and
, , , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
2.3. Semen Collection and Evaluation
2.4. Bacterial Culture, Identification, and Quantification
2.5. DNA Extraction, PCR Amplification, and Bioinformatics Analysis
2.6. Statistical Analysis
3. Results
3.1. Semen Characteristics and Total Bacterial Count across Semen Samples
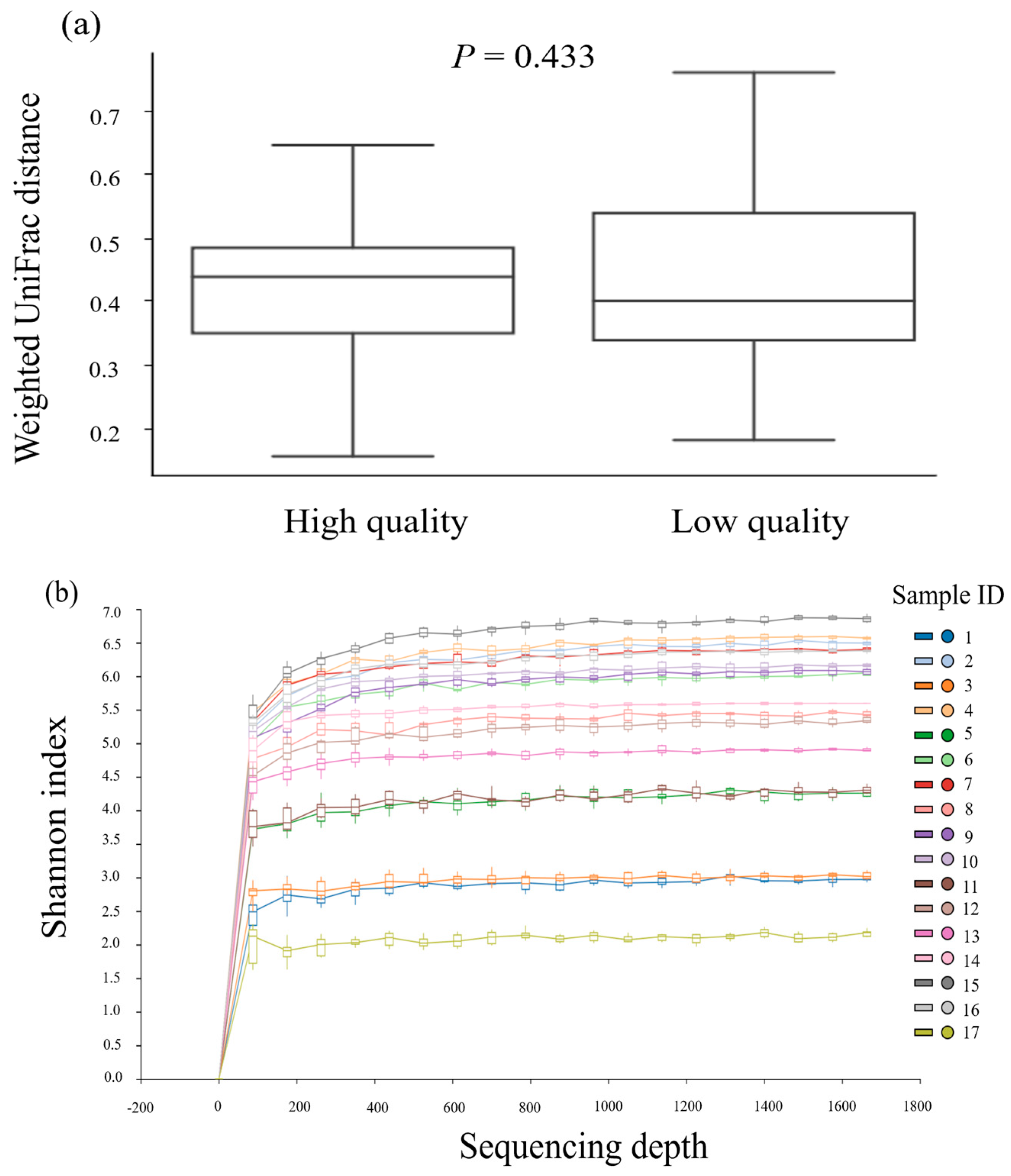
3.2. The Richness and Diversity across Semen Samples
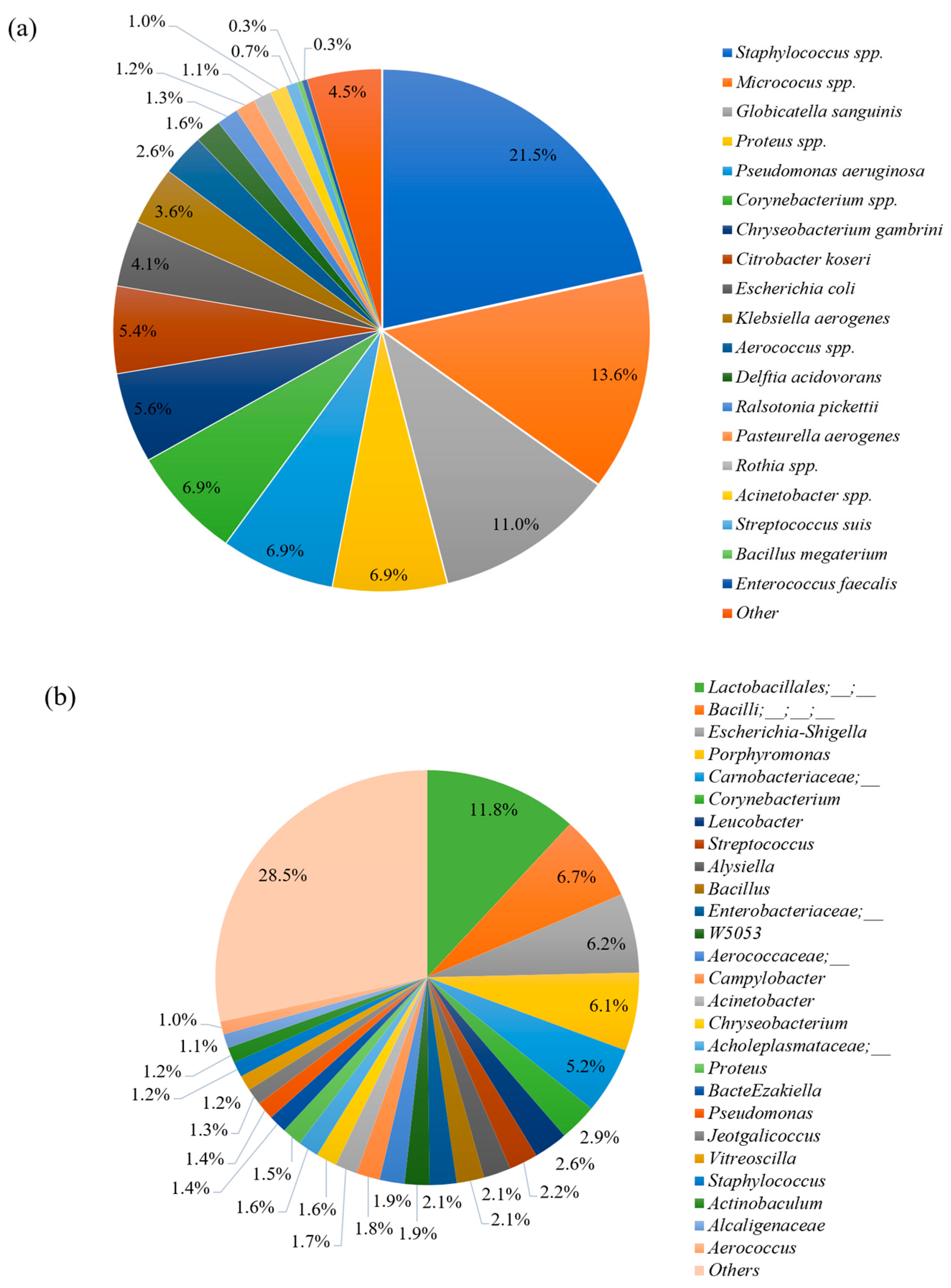
3.3. Bacteria Identification Using Bacterial Culture and Next-Generation Sequencing Methods
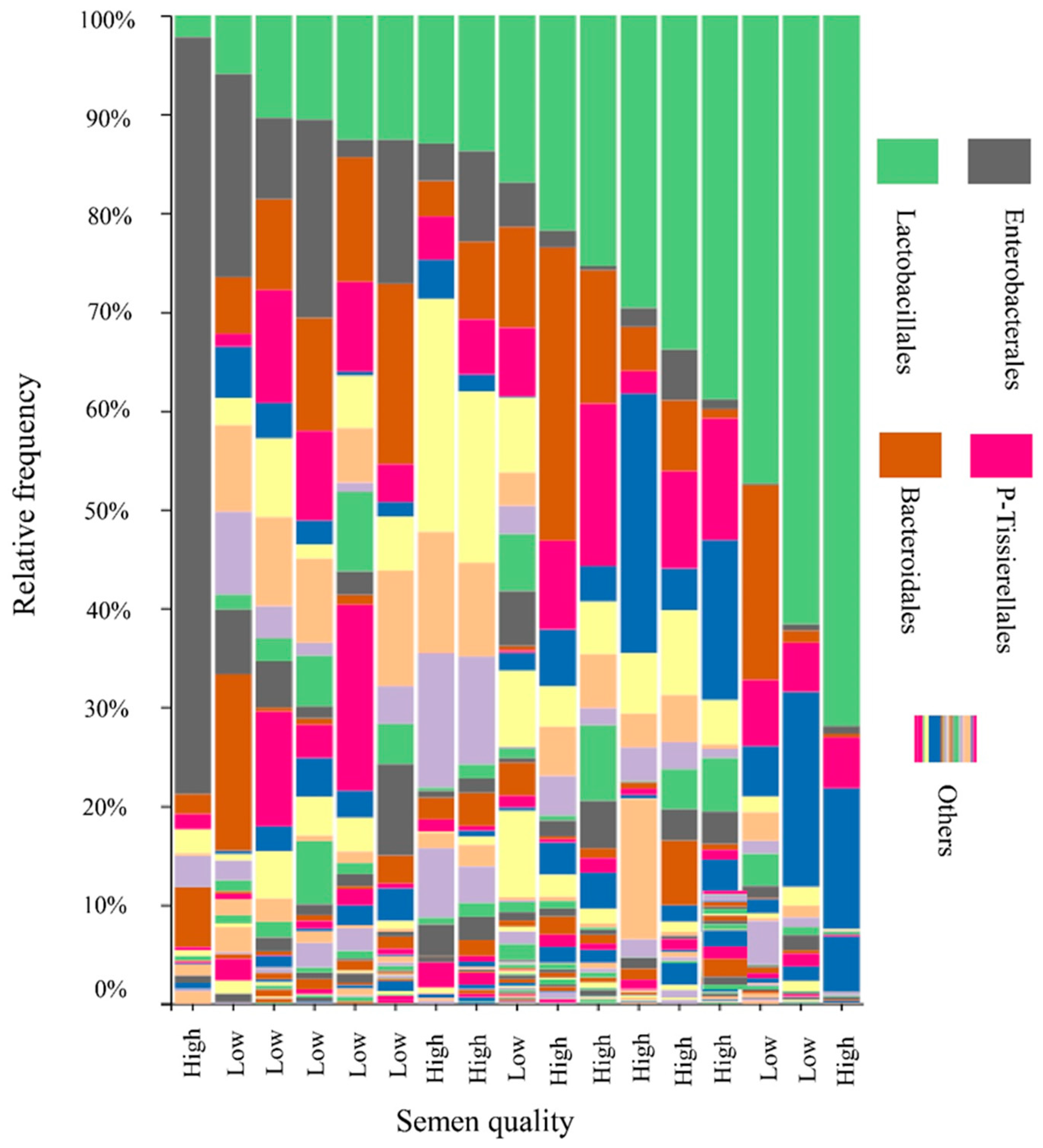
3.4. Boar Seminal Microbiota Profile in Relation to Semen Quality
3.5. Correlation between Major Bacteria Detected by Next-Generation Sequencing Method
4. Discussion
4.1. Boar Seminal Microbiota
4.2. Boar Seminal Microbial Richness and Diversity
4.3. Differences in the Bacteria Detected by Different Techniques
4.4. Association between Bacteria and Boar Semen Qualities
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Althouse, G.C.; Kuster, C.E.; Clark, S.G.; Weisiger, R.M. Field investigations of bacterial contaminants and their effects on extended porcine semen. Theriogenology 2000, 53, 1167–1176. [Google Scholar] [CrossRef]
- Althouse, G.C.; Lu, K.G. Bacteriospermia in extended porcine semen. Theriogenology 2005, 63, 573–584. [Google Scholar] [CrossRef]
- Ngo, C.B.; Suwimonteerabut, J.; Prapasarakul, N.; Morrell, J.M.; Tummaruk, P. Bacteriospermia and its antimicrobial resistance in relation to boar sperm quality during short-term storage with or without antibiotics in a tropical environment. Porc. Health Manag. 2023, 9, 21. [Google Scholar] [CrossRef]
- Gòdia, M.; Ramayo-Caldas, Y.; Zingaretti, L.M.; Darwich, L.; López, S.; Rodríguez-Gil, J.E.; Yeste, M.; Sanchez, A.; Clop, A. A pilot RNA-seq study in 40 pietrain ejaculates to characterize the porcine sperm microbiome. Theriogenology 2020, 157, 525–533. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.; Yang, Q.; Li, P.; Wen, Y.; Han, X.; Li, B.; Jiang, H.; Li, X. Genomic sequencing reveals the diversity of seminal bacteria and relationships to reproductive potential in boar sperm. Front. Microbiol. 2020, 11, 1873. [Google Scholar] [CrossRef]
- Hou, D.; Zhou, X.; Zhong, X.; Settles, M.L.; Herring, J.; Wang, L.; Abdo, Z.; Forney, L.J.; Xu, C. Microbiota of the seminal fluid from healthy and infertile men. Fertil. Steril. 2013, 100, 1261–1269. [Google Scholar] [CrossRef]
- Baud, D.; Pattaroni, C.; Vulliemoz, N.; Castella, V.; Marsland, B.J.; Stojanov, M. Sperm microbiota and its impact on semen parameters. Front. Microbiol. 2019, 10, 234. [Google Scholar] [CrossRef]
- Niederwerder, M.; Jaing, C.; Thissen, J.; Cino-Ozuna, A.; McLoughlin, K.; Rowland, R. Microbiome associations in pigs with the best and worst clinical outcomes following co-infection with porcine reproductive and respiratory syndrome virus (PRRSV) and porcine circovirus type 2 (PCV2). Vet. Microbiol. 2016, 188, 1–11. [Google Scholar] [CrossRef]
- Niu, Q.; Li, P.; Hao, S.; Kim, S.W.; Du, T.; Hua, J.; Huang, R. Characteristics of gut microbiota in sows and their relationship with apparent nutrient digestibility. Int. J. Mol. Sci. 2019, 20, 870. [Google Scholar] [CrossRef]
- Monteiro, M.S.; Poor, A.P.; Muro, B.B.D.; Carnevale, R.F.; Leal, D.F.; Garbossa, C.A.P.; Moreno, A.M.; Almond, G. The sow microbiome: Current and future perspectives to maximize the productivity in swine herds. J. Swine Health Prod. 2022, 30, 238–250. [Google Scholar] [CrossRef]
- Weng, S.L.; Chiu, C.M.; Lin, F.M.; Huang, W.C.; Liang, C.; Yang, T.; Liu, C.Y.; Wu, W.Y.; Chang, Y.A.; Chang, T.H.; et al. Bacterial communities in semen from men of infertile couples: Metagenomic sequencing reveals relationships of seminal microbiota to semen quality. PLoS ONE 2014, 9, e110152. [Google Scholar] [CrossRef] [PubMed]
- Sanglard, L.P.; Schmitz-Esser, S.; Gray, K.A.; Linhares, D.C.L.; Yeoman, C.J.; Dekkers, J.C.M.; Neiderwerder, M.C.; Serao, N.V.L. Vaginal microbiota diverges in sows with low and high reproductive performance after porcine reproductive and respiratory syndrome vaccination. Sci. Rep. 2020, 10, 3046. [Google Scholar] [CrossRef] [PubMed]
- Gączarzewicz, D.; Udała, J.; Piasecka, M.; Błaszczyk, B.; Stankiewicz, T. Bacterial contamination of boar semen and its relationship to sperm quality preserved in commercial extender containing gentamicin sulfate. Pol. J. Vet. Sci. 2016, 19, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Kuster, C.E.; Althouse, G.C. The impact of bacteriospermia on boar sperm storage and reproductive performance. Theriogenology 2016, 85, 21–26. [Google Scholar] [CrossRef]
- Sepúlveda, L.; Bussalleu, E.; Yeste, M.; Bonet, S. Effect of Pseudomonas aeruginosa on sperm capacitation and protein phosphorylation of boar spermatozoa. Theriogenology 2016, 85, 1421–1431. [Google Scholar] [CrossRef]
- Mulder, I.E.; Schmidt, B.; Stokes, C.R.; Lewis, M.; Bailey, M.; Aminov, R.I.; Prosser, J.I.; Gill, B.P.; Pluske, J.R.; Mayer, C.D.; et al. Environmentally acquired bacteria influence microbial diversity and natural innate immune responses at gut surfaces. BMC Biol. 2009, 7, 79. [Google Scholar] [CrossRef]
- Viudes-de-Castro, M.P.; Marco-Jimenez, F.; Vicente, J.S.; Marin, C. Antibacterial activity of some molecules added to rabbit semen extender as alternative to antibiotics. Animals 2021, 11, 1178. [Google Scholar] [CrossRef]
- Lugar, D.W.; Harlow, K.E.; Hundley, J.; Goncalves, M.; Bergstrom, J.; Stewart, K.R. Effects of increased levels of supplemental vitamins during the summer in a commercial artificial insemination boar stud. Animal 2019, 13, 2556–2568. [Google Scholar] [CrossRef]
- Ngo, C.B.; Sooksong, S.; Am-in, N.; Tummaruk, P. Semen production capacity among boar breeds and effect of organic antioxidant supplementation on sperm characteristics during the hot season in Thailand. Thai J. Vet. Med. 2022, 52, 441–449. [Google Scholar] [CrossRef]
- Tretipskul, C.; Am-in, N.; Tummaruk, P.; Techakumphu, M. Season and breed effects on sperm production in PRRS free boars. Thai J. Vet. Med. 2013, 42, 469–476. [Google Scholar] [CrossRef]
- Rungruangsak, J.; Sangkaphet, S.; Buranaamnuay, K.; Pongpeng, P.; Tummaruk, P. Boar sperm production in a tropical environment. Thai J. Vet. Med. 2021, 51, 213–220. [Google Scholar] [CrossRef]
- Martin, N.C.; Pirie, A.A.; Ford, L.V.; Callaghan, C.L.; McTurk, K.; Lucy, D.; Scrimger, D.G. The use of phosphate buffered saline for the recovery of cells and spermatozoa from swabs. Sci. Justice 2006, 46, 179–184. [Google Scholar] [CrossRef]
- Suwimonteerabutr, J.; Chumsri, S.; Tummaruk, P.; Nuntapaitoon, M. Butaphosphan and cyanocobalamin supplementation in semen extender on chilled boar sperm quality and life span. Front. Vet. Sci. 2020, 7, 592162. [Google Scholar] [CrossRef] [PubMed]
- Martín, L.O.; Muñoz, E.C.; De Cupere, F.; Van Driessche, E.; Echemendia-Blanco, D.; Rodríguez, J.M.; Beeckmans, S. Bacterial contamination of boar semen affects the litter size. Anim. Reprod. Sci. 2010, 120, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Akhter, S.; Ansari, M.S.; Andrabi, S.; Ullah, N.; Qayyum, M. Effect of antibiotics in extender on bacterial and spermatozoal quality of cooled buffalo (Bubalus bubalis) bull semen. Reprod. Domest. Anim. 2008, 43, 272–278. [Google Scholar] [CrossRef]
- Bailey, D.; Diamandis, E.; Greub, G.; Poutanen, S.; Christensen, J.J.; Kostrzew, M. Use of MALDI-TOF for diagnosis of microbial infections. Clin. Chem. 2013, 59, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef]
- Alcolea-Medina, A.; Mtc, F.; Montiel, N.; Mpl, G.; Cd, S.; North, N.; Lirola, M.J.M.; Wilks, M. An improved simple method for the identification of mycobacteria by MALDI-TOF MS (matrix-assisted laser desorption-ionization mass spectrometry). Sci. Rep. 2019, 9, 20216. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 2018, 3, e00021-18. [Google Scholar] [CrossRef] [PubMed]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Hamady, M.; Lozupone, C.; Knight, R. Fast UniFrac: Facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010, 4, 17–27. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Dillon, M.R.; Zhang, Y.; Rideout, J.R.; Bolyen, E.; Li, H.; Albert, P.S.; Caporaso, J.G. q2-longitudinal: Longitudinal and paired-sample analyses of microbiome data. mSystems 2018, 3, e00219-18. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef]
- Kraemer, J.G.; Ramette, A.; Aebi, S.; Oppliger, A.; Hilty, M. Influence of pig farming on the human nasal microbiota: Key role of airborne microbial communities. Appl. Environ. Microbiol. 2018, 84, 6. [Google Scholar] [CrossRef]
- Johnson, T.A.; Looft, T.; Severin, A.J.; Bayles, D.O.; Nasko, D.J.; Wommack, K.E.; Howe, A.; Allen, H.K. The in-feed antibiotic carbadox induces phage gene transcription in the swine gut microbiome. mBio 2017, 8, e00709-17. [Google Scholar] [CrossRef]
- Makarova, K.; Slesarev, A.; Wolf, Y.; Sorokin, A.; Mirkin, B.; Koonin, E. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 15611–15616. [Google Scholar] [CrossRef] [PubMed]
- Bukharin, O.V.; Kuz’min, M.D.; Ivanov Iu, B. The role of the microbial factor in the pathogenesis of male infertility. Zhurnal Mikrobiol. Epidemiol. Immunobiol. 2000, 3, 106–110. [Google Scholar]
- Ivanov, I.B.; Kuzmin, M.D.; Gritsenko, V.A. Microflora of the seminal fluid of healthy men and men suffering from chronic prostatitis syndrome. Int. J. Androl. 2009, 32, 462–467. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Guevarra, R.B.; Hong, S.H.; Cho, J.H.; Kim, B.R.; Shin, J.; Lee, J.H.; Kang, B.N.; Kim, Y.H.; Wattanaphansak, S.; Isaccson, R.E.; et al. The dynamics of the piglet gut microbiome during the weaning transition in association with health and nutrition. J. Anim. Sci. Biotechnol. 2018, 9, 54. [Google Scholar] [CrossRef]
- Pinart, E.; Domènech, E.; Bussalleu, E.; Yeste, M.; Bonet, S. A comparative study of the effects of Escherichia coli and Clostridium perfringens upon boar semen preserved in liquid storage. Anim. Reprod. Sci. 2017, 177, 65–78. [Google Scholar] [CrossRef]
- Ohkubo, T.; Matsumoto, Y.; Cho, O.; Ogasawara, Y.; Sugita, T. Delftia acidovorans secretes substances that inhibit the growth of Staphylococcus epidermidis through TCA cycle-triggered ROS production. PLoS ONE 2021, 16, e0253618. [Google Scholar] [CrossRef]
- Rungruangsak, J.; Suwimonteerabutr, J.; Buranaamnuay, K.; Chankrachang, A.; Tummaruk, P. A comparative study of two methods to determine acrosome integrity of frozen-thawed boar sperm: FITC-PNA/EthD-1 versus Coomassie blue staining. Vet. Stanica 2021, 52, 629–638. [Google Scholar] [CrossRef]
- Santos, C.S.; Silva, A.M.; Maia, K.M.; Rodrigues, G.S.O.; Feijó, F.M.C.; Alves, N.D.; Oliveira, M.F.; Silva, A.R. Composition of semen and foreskin mucosa aerobic microbiota and its impact on sperm parameters of captive collared peccaries (Pecari tajacu). J. Appl. Microbiol. 2020, 129, 521–531. [Google Scholar] [CrossRef]
- Xu, S.; Dong, Y.; Shi, J.; Li, Z.; Che, L.; Lin, Y.; Li, J.; Feng, B.; Fang, Z.; Yong, Z.; et al. Responses of vaginal microbiota to dietary supplementation with lysozyme and its relationship with rectal microbiota and sow performance from late gestation to early lactation. Animals 2021, 11, 593. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Group | p Value | |
|---|---|---|---|
| Low Quality (n = 8) | High Quality (n = 9) | ||
| Volume (mL) | 170 ± 29.8 | 215 ± 31.6 | 0.320 |
| Concentration (×106 sperm/mL) | 345.0 ± 38.6 | 357 ± 35.9 | 0.321 |
| Total sperm per ejaculate (×109 sperm) | 59.9 ± 9.4 | 54.2 ± 8.8 | 0.662 |
| pH | 7.6 ± 0.1 | 7.4 ± 0.1 | 0.445 |
| Total sperm motility (%) | 67.4 ± 2.8 a | 81.7 ± 2.7 b | 0.002 |
| - Progressive motility (%) | 56.0 ± 3.1 a | 74.6 ± 2.9 b | <0.001 |
| - Non-motile sperm (%) | 32.6 ± 2.8 a | 18.3 ± 2.7 b | 0.002 |
| - Linear velocity (VSL, µm/s) | 26.6 ± 2.0 | 31.5 ± 1.9 | 0.098 |
| - Curvilinear velocity (VCL, µm/s) | 87.1 ± 4.0 | 94.1 ± 3.8 | 0.218 |
| - Average path velocity (VAP, µm/s) | 46.0 ± 2.8 | 51.2 ± 2.6 | 0.188 |
| - Linearity (a ratio of VSL/VCL; LIN, %) | 29.6 ± 2.1 | 35.1 ± 2.0 | 0.080 |
| - Straightness (a ratio of VSL/VAP; STR, %) | 53.6 ± 2.1 a | 60.5 ± 2.1 b | 0.028 |
| - Wobbe (a ratio of VAP/VCL; WOB, %) | 51.2 ± 1.8 | 54.4 ± 1.7 | 0.204 |
| Sperm viability (%) | 71.3 ± 1.3 a | 83.6 ± 1.2 b | <0.001 |
| Sperm acrosome integrity (%) | 80.7 ± 2.0 | 86.8 ± 2.0 | 0.050 |
| Sperm membrane functionality (%) | 58.3 ± 2.4 a | 72.6 ± 2.2 b | <0.001 |
| Sperm mitochondrial activity (%) | 67.4 ± 2.9 a | 79.8 ± 2.7 b | 0.007 |
| Total bacterial count (CFU/mL, log10) | 4.4 ± 0.2 | 3.9 ± 0.2 | 0.065 |
| Bacterial Count (CFU/mL, log10) | Correlation Coefficient | |||||
|---|---|---|---|---|---|---|
| pH | Motility | Viability | Acrosome Integrity | Sperm Membrane Functionality | Sperm Mitochondrial Activity | |
| Total bacterial count | 0.631 ** | NS | −0.495 * | NS | −0.707 *** | NS |
| Staphylococcus spp. | 0.576 * | NS | NS | NS | −0.577 * | NS |
| Micrococcus spp. | NS | NS | NS | NS | −0.529 * | NS |
| Globicatella sanguinis | NS | NS | NS | −0.699 ** | −0.534 * | −0.507 * |
| Pseudomonas aeruginosa | NS | NS | NS | NS | NS | NS |
| Proteus spp. | NS | NS | NS | −0.581 * | NS | NS |
| Corynebacterium spp. | 0.476 * | NS | NS | NS | NS | NS |
| Chryseobacterium gambrini | NS | NS | NS | NS | NS | NS |
| Citrobacter koseri | NS | NS | NS | NS | NS | NS |
| Escherichia coli | 0.706 *** | NS | NS | NS | NS | NS |
| Klebsiella aerogenes | NS | NS | NS | NS | NS | NS |
| Pasteurella aerogenes | NS | NS | NS | NS | NS | NS |
| Rothia spp. | NS | NS | NS | NS | NS | NS |
| Delftia acidovorans | NS | 0.634 *** | 0.644 *** | 0.500 * | 0.661 ** | 0.512 * |
| Bacteria | Group | p Value | |
|---|---|---|---|
| Low Quality | High Quality | ||
| Significant difference | |||
| Alysiella | 11.6 ± 10.4 a | 6.7 ± 10.4 b | 0.048 |
| Myroides | 7.0 ± 7.7 a | 10.8 ± 7.7 b | 0.045 |
| Bacilli | 6.3 ± 10.4 a | 11.4 ± 10.4 b | 0.043 |
| Non-significant differences | |||
| Acholeplasmataceae | 7.3 ± 10.3 | 10.5 ± 10.3 | NS |
| Acinetobacter | 7.7 ± 10.4 | 10.1 ± 10.4 | NS |
| Actinobaculum | 8.0 ± 10.4 | 9.8 ± 10.4 | NS |
| Aerococcaceae | 9.4 ± 10.4 | 9.4 ± 10.4 | NS |
| Aerococcus | 10.5 ± 10.4 | 7.6± 10.4 | NS |
| Alcaligenaceae | 10.3 ± 10.4 | 7.8 ± 10.4 | NS |
| Bacillus | 10.5 ± 9.3 | 7.6 ± 9.3 | NS |
| Bacteroides | 8.5 ± 10.3 | 9.4 ± 10.3 | NS |
| Campylobacter | 11.0 ± 10.4 | 7.2 ± 10.4 | NS |
| Carnobacteriaceae | 6.8 ± 10.4 | 10.9 ± 10.4 | NS |
| Chryseobacterium | 9.3 ± 10.3 | 8.7 ± 10.3 | NS |
| Clostridium sensu stricto 1 | 10.4 ± 10.3 | 7.8 ± 10.3 | NS |
| Corynebacterium | 10.3 ± 10.4 | 7.9 ± 10.4 | NS |
| Enterobacteriaceae | 8.7 ± 10.2 | 9.3 ± 10.2 | NS |
| Escherichia–shigella | 9.9 ± 10.3 | 8.2 ± 10.3 | NS |
| Ezakiella | 10.2 ± 10.3 | 7.9 ± 10.3 | NS |
| Flavobacterium | 10.1 ± 9.8 | 8.0 ± 9.8 | NS |
| Globicatella | 10.4 ± 10.3 | 7.8 ± 10.4 | NS |
| Jeotgalicoccus | 10.4 ± 10.4 | 7.7 ± 10.4 | NS |
| Lactobacillales | 7.6 ± 10.4 | 10.3 ± 10.4 | NS |
| Lactobacillus | 10.0 ± 10.3 | 8.1 ± 10.3 | NS |
| Leucobacter | 7.5 ± 6.9 | 10.3 ± 6.9 | NS |
| Micrococcus | 9.9 ± 10.3 | 8.2 ± 10.3 | NS |
| Mobiluncus | 11.4 ± 10.3 | 6.8 ± 10.4 | NS |
| Pasteurella | 10.3 ± 8.9 | 7.9 ± 8.9 | NS |
| Porphyromonas | 10.3 ± 10.4 | 7.8 ± 10.4 | NS |
| Prevotella | 10.1 ± 10.3 | 8.0 ± 10.3 | NS |
| Proteus | 11.2 ± 10.3 | 7.0 ± 10.3 | NS |
| Pseudomonas | 7.8 ± 10.3 | 10.5 ± 10.3 | NS |
| Rothia | 10.9 ± 10.3 | 7.3 ± 10. 3 | NS |
| Staphylococcus | 9.5 ± 10.3 | 8.6 ± 10.3 | NS |
| Streptococcus | 9.4 ± 10.4 | 8.6 ± 10.4 | NS |
| W5053 | 8.9 ± 10.4 | 9.1 ± 10.4 | NS |
| Bacteria | Correlation Coefficient | ||||
|---|---|---|---|---|---|
| Alysiella | Bacilli | Escherichia–Shigella | Myroides | Prophymonas | |
| Aerococcaceae | NS | 0.533 * | −0.506 * | −0.614 ** | NS |
| Bacillus | NS | NS | NS | NS | 0.705 ** |
| Campylobacter | 0.714 ** | NS | NS | −0.486 * | NS |
| Chryseobacterium | NS | −0.498 * | 0.611 ** | NS | NS |
| Corynebacterium | 0.723 ** | NS | NS | NS | NS |
| Globicatella | NS | NS | NS | −0.503 * | NS |
| Lactobacillales | NS | 0.796 *** | −0.754 *** | NS | NS |
| Lactobacillus | 0.485 * | NS | NS | NS | NS |
| Leucobacter | NS | NS | NS | 0.894 *** | NS |
| Prevotella | 0.622 ** | NS | NS | NS | 0.587 * |
| Proteus | NS | −0.520 * | 0.533 * | −0.495 * | NS |
| Pseudomonas | NS | NS | NS | 0.704 ** | NS |
| Rothia | NS | −0.740 *** | 0.673 ** | NS | NS |
| Staphylococcus | 0.489 * | NS | NS | NS | NS |
| Streptococcus | 0.547 * | NS | NS | NS | 0.578 * |
| Vitreoscilla | NS | NS | NS | 0.098 *** | NS |
| W5053 | NS | 0.484 * | −0.597 * | NS | NS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngo, C.; Suwimonteerabutr, J.; Apiwatsiri, P.; Saenkankam, I.; Prapasarakul, N.; Morrell, J.M.; Tummaruk, P. Boar Seminal Microbiota in Relation to Sperm Quality under Tropical Environments. Animals 2023, 13, 3837. https://doi.org/10.3390/ani13243837
Ngo C, Suwimonteerabutr J, Apiwatsiri P, Saenkankam I, Prapasarakul N, Morrell JM, Tummaruk P. Boar Seminal Microbiota in Relation to Sperm Quality under Tropical Environments. Animals. 2023; 13(24):3837. https://doi.org/10.3390/ani13243837
Chicago/Turabian StyleNgo, CongBang, Junpen Suwimonteerabutr, Prasert Apiwatsiri, Imporn Saenkankam, Nuvee Prapasarakul, Jane M. Morrell, and Padet Tummaruk. 2023. "Boar Seminal Microbiota in Relation to Sperm Quality under Tropical Environments" Animals 13, no. 24: 3837. https://doi.org/10.3390/ani13243837
APA StyleNgo, C., Suwimonteerabutr, J., Apiwatsiri, P., Saenkankam, I., Prapasarakul, N., Morrell, J. M., & Tummaruk, P. (2023). Boar Seminal Microbiota in Relation to Sperm Quality under Tropical Environments. Animals, 13(24), 3837. https://doi.org/10.3390/ani13243837