
Conflicting Evidence between Clinical Perception and Molecular Epidemiology: The Case of Fowl Adenovirus D
 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Dataset Preparation
2.2. Phylodynamic and Phylogeographic Analysis
2.3. Selective Pressure Analysis
2.4. Homology Modeling
3. Results
3.1. Dataset
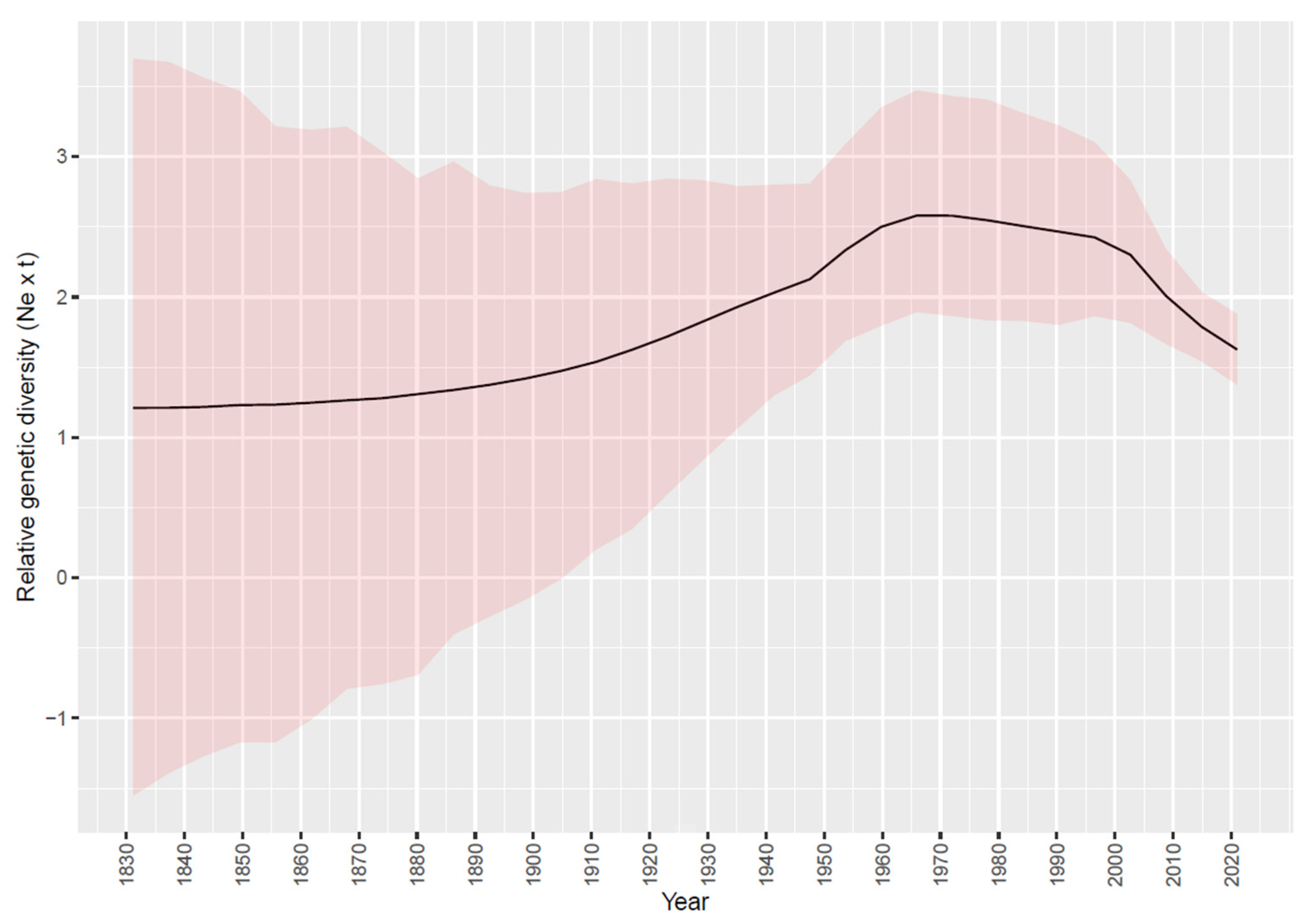
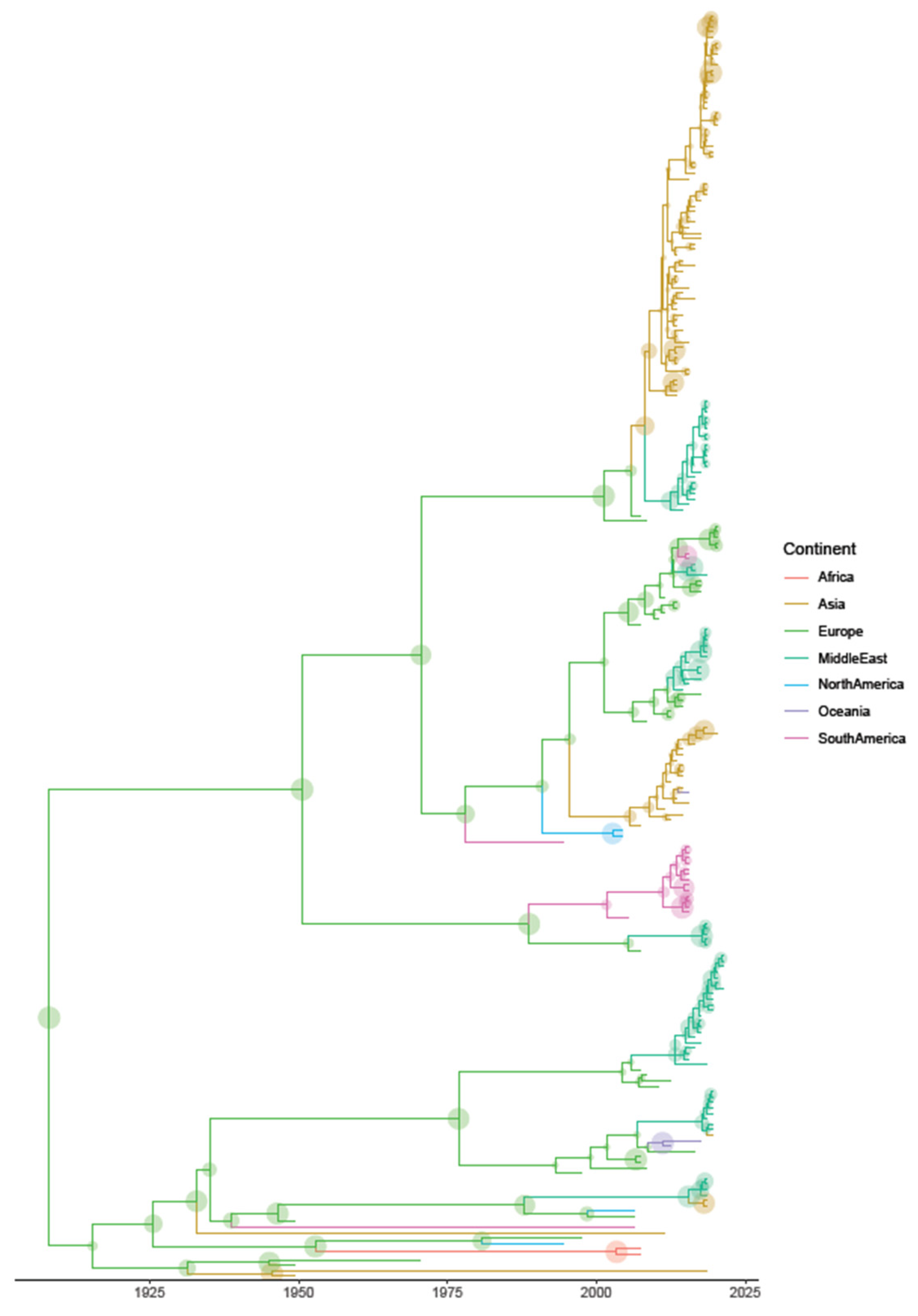
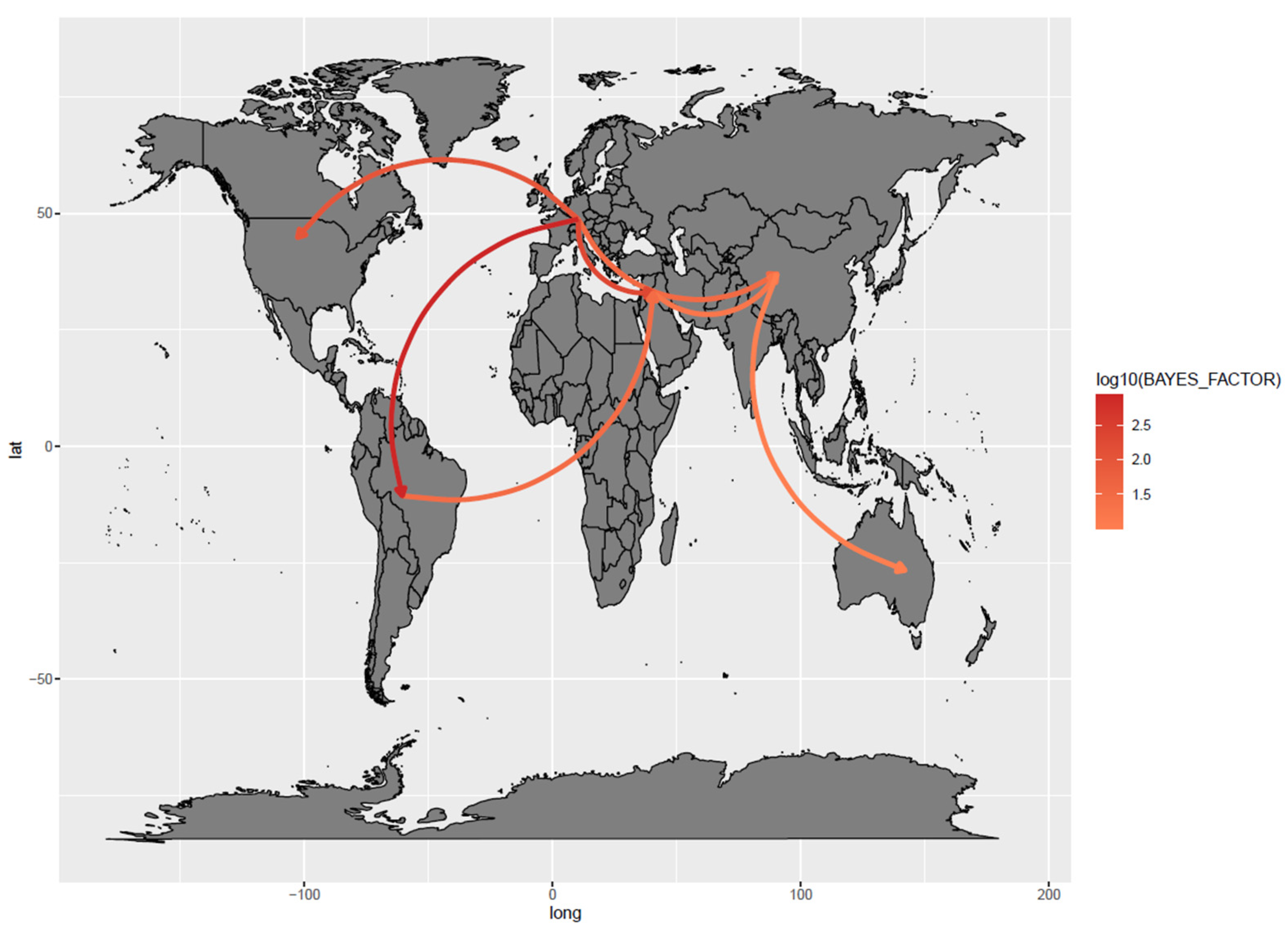
3.2. Phylodynamic Analysis
3.3. Selective Forces
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hess, M. Aviadenovirus Infections. In Diseases of Poultry, 13th ed.; Glisson, J.R., McDougald, L.R., Nolan, L.K., Suarez, D.L., Nair, V.L., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; pp. 290–300. [Google Scholar]
- Marek, A.; Günes, A.; Schulz, E.; Hess, M. Classification of Fowl Adenoviruses by Use of Phylogenetic Analysis and High-Resolution Melting-Curve Analysis of the Hexon L1 Gene Region. J. Virol. Methods 2010, 170, 147–154. [Google Scholar] [CrossRef]
- Bertran, K.; Blanco, A.; Antilles, N.; Nofrarías, M.; Valle, R.M.; Cobos, À.; Ramis, A.; Biarnés, M.; Majó, N. A 10-Year Retrospective Study of Inclusion Body Hepatitis in Meat-Type Chickens in Spain (2011–2021). Viruses 2021, 13, 2170. [Google Scholar] [CrossRef]
- Liu, J.; Mei, N.; Wang, Y.; Shi, X.; Chen, H. Identification of a Novel Immunological Epitope on Hexon of Fowl Adenovirus Serotype 4. AMB Express 2021, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Schachner, A.; Matos, M.; Grafl, B.; Hess, M. Fowl Adenovirus-Induced Diseases and Strategies for Their Control–a Review on the Current Global Situation. Avian Pathol. 2018, 47, 111–126. [Google Scholar] [CrossRef]
- Hess, M. Commensal or Pathogen–a Challenge to Fulfil Koch’s Postulates. Br. Poult. Sci. 2017, 58, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, S.D.; Rautenschlein, S.; Mahsoub, H.M.; Pierson, F.W.; Reed, W.M.; Jack, S.W. Adenovirus Infections. In Diseases of Poultry; Wiley: Hoboken, NJ, USA, 2019; pp. 321–363. [Google Scholar] [CrossRef]
- Oliver-Ferrando, S.; Dolz, R.; Calderón, C.; Valle, R.; Rivas, R.; Pérez, M.; Biarnés, M.; Blanco, A.; Bertran, K.; Ramis, A.; et al. Epidemiological and Pathological Investigation of Fowl Aviadenovirus Serotypes 8b and 11 Isolated from Chickens with Inclusion Body Hepatitis in Spain (2011–2013). Avian Pathol. 2017, 46, 157–165. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, D.; Nuñez, L.F.N.; Santander Parra, S.H.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Molecular Characterization of Fowl Adenovirus Group I in Commercial Broiler Chickens in Brazil. Virusdisease 2018, 29, 83. [Google Scholar] [CrossRef]
- Chitradevi, S.; Sukumar, K.; Suresh, P.; Balasubramaniam, G.A.; Kannan, D. Molecular Typing and Pathogenicity Assessment of Fowl Adenovirus Associated with Inclusion Body Hepatitis in Chicken from India. Trop. Anim. Health Prod. 2021, 53, 1–12. [Google Scholar] [CrossRef]
- Niczyporuk, J.S.; Kozdrun, W.; Czekaj, H.; Piekarska, K.; Stys-Fijol, N. Characterisation of Adenovirus Strains Represented Species B and E Isolated from Broiler Chicken Flocks in Eastern Poland. Heliyon 2021, 7, e06225. [Google Scholar] [CrossRef]
- Mase, M.; Hiramatsu, K.; Nishijima, N.; Iguchi, H.; Honda, S.; Hanyu, S.; Iseki, H.; Watanabe, S. Fowl Adenoviruses Type 8b Isolated from Chickens with Inclusion Body Hepatitis in Japan. Avian Dis. 2020, 64, 330–334. [Google Scholar] [CrossRef]
- Radwan, M.M.; El-Deeb, A.H.; Mousa, M.R.; El-Sanousi, A.A.; Shalaby, M.A. First Report of Fowl Adenovirus 8a from Commercial Broiler Chickens in Egypt: Molecular Characterization and Pathogenicity. Poult. Sci. 2019, 98, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Prentza, Z.; Paparounis, T.; Tsiouris, V.; Centonze, G.; Legnardi, M.; Catelli, E.; Tucciarone, C.M.; Koutoulis, K.; Cecchinato, M. Molecular Epidemiology of Fowl Adenoviruses in Greece. Poult. Sci. 2020, 99, 5983. [Google Scholar] [CrossRef]
- Schachner, A.; Grafl, B.; Hess, M. Spotlight on Avian Pathology: Fowl Adenovirus (FAdV) in Chickens and beyond—An Unresolved Host-Pathogen Interplay. Avian Pathol. 2020, 50, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Atkins, K.E.; Read, A.F.; Savill, N.J.; Renz, K.G.; Islam, A.F.; Walkden-Brown, S.W.; Woolhouse, M.E.J. Vaccination and reduced cohort duration can drive virulence evolution: Marek’s disease virus and industrialized agriculture. Evolution 2013, 67, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Tatem, A.J.; Lemey, P. Virus Evolution and Transmission in an Ever More Connected World. Proc. Biol. Sci. 2015, 282, 20142878. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, T.; Bai, Y.; Li, H.; Wang, Y.; Bi, Y.; Chang, J.; Xu, B. Live Poultry Trading Drives China’s H7N9 Viral Evolution and Geographical Network Propagation. Front. Public Health 2018, 6, 210. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, X.; Lemey, P.; Suchard, M.A.; Bi, Y.; Shi, W.; Liu, D.; Qi, W.; Zhang, G.; Stenseth, N.C.; et al. Assessing the Role of Live Poultry Trade in Community-Structured Transmission of Avian Influenza in China. Proc. Natl. Acad. Sci. USA 2020, 117, 5949–5954. [Google Scholar] [CrossRef]
- Hilborn, R.; Banobi, J.; Hall, S.J.; Pucylowski, T.; Walsworth, T.E. The Environmental Cost of Animal Source Foods. Front Ecol Environ 2018, 16, 329–335. [Google Scholar] [CrossRef]
- Fox, M.A. Deep Vegetarianism; Temple University Press: Philadelphia, PA, USA, 1999. [Google Scholar]
- Izmirli, S.; Phillips, C.J.C. The Relationship between Student Consumption of Animal Products and Attitudes to Animals in Europe and Asia. Br. Food J. 2011, 113, 436–450. [Google Scholar] [CrossRef]
- Msami, D.H. Poultry Sector Country Review; The Food and Agriculture Organization (FAO): Rome, Italy, 2008. [Google Scholar]
- Wilson, R.T. Poultry Production and Performance in the Federal Democratic Republic of Ethiopia. World’s Poult. Sci. J. 2010, 66, 441–453. [Google Scholar] [CrossRef]
- Standley, K. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. (Outlines Version 7). Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A Genetic Algorithm for Recombination Detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis Testing Using Phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Helmboldt, C.F.; Frazier, M.N. Avian Hepatic Inclusion Bodies of Unknown Significance. Avian Dis. 1963, 7, 446–450. [Google Scholar] [CrossRef]
- Athukorala, A.; Helbig, K.J.; Mcsharry, B.P.; Forwood, J.K.; Sarker, S. Adenoviruses in Avian Hosts: Recent Discoveries Shed New Light on Adenovirus Diversity and Evolution. Viruses 2022, 14, 1767. [Google Scholar] [CrossRef]
- Fleming-Davies, A.E.; Dukic, V.; Andreasen, V.; Dwyer, G. Effects of Host Heterogeneity on Pathogen Diversity and Evolution. Ecol. Lett. 2015, 18, 1252. [Google Scholar] [CrossRef] [PubMed]
- Firth, C.; Kitchen, A.; Shapiro, B.; Suchard, M.A.; Holmes, E.C.; Rambaut, A. Using Time-Structured Data to Estimate Evolutionary Rates of Double-Stranded DNA Viruses. Mol. Biol. Evol. 2010, 27, 2038–2051. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Katzourakis, A. Time-Dependent Rate Phenomenon in Viruses. J. Virol. 2016, 90, 7184–7195. [Google Scholar] [CrossRef]
- Ho, S.Y.W.; Lanfear, R.; Bromham, L.; Phillips, M.J.; Soubrier, J.; Rodrigo, A.G.; Cooper, A. Time-Dependent Rates of Molecular Evolution. Mol. Ecol. 2011, 20, 3087–3101. [Google Scholar] [CrossRef] [PubMed]
- Duchêne, S.; Holmes, E.C.; Ho, S.Y.W. Analyses of Evolutionary Dynamics in Viruses Are Hindered by a Time-Dependent Bias in Rate Estimates. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140732. [Google Scholar] [CrossRef]
- Franzo, G.; Cecchinato, M.; Tosi, G.; Fiorentini, L.; Faccin, F.; Tucciarone, C.M.; Trogu, T.; Barbieri, I.; Massi, P.; Moreno, A. GI-16 Lineage (624/I or Q1), There and Back Again: The History of One of the Major Threats for Poultry Farming of Our Era. PLoS ONE 2018, 13, e0203513. [Google Scholar] [CrossRef]
- Franzo, G.; He, W.; Correa-Fiz, F.; Li, G.; Legnardi, M.; Su, S.; Segalés, J. A Shift in Porcine Circovirus 3 (PCV-3) History Paradigm: Phylodynamic Analyses Reveal an Ancient Origin and Prolonged Undetected Circulation in the Worldwide Swine Population. Adv. Sci. 2019, 6, 1901004. [Google Scholar] [CrossRef]
- Franzo, G.; Cortey, M.; Segalés, J.; Hughes, J.; Drigo, M. Phylodynamic Analysis of Porcine Circovirus Type 2 Reveals Global Waves of Emerging Genotypes and the Circulation of Recombinant Forms. Mol. Phylogenetics Evol. 2016, 100, 269–280. [Google Scholar] [CrossRef]
- Franzo, G.; Massi, P.; Tucciarone, C.M.; Barbieri, I.; Tosi, G.; Fiorentini, L.; Ciccozzi, M.; Lavazza, A.; Cecchinato, M.; Moreno, A. Think Globally, Act Locally: Phylodynamic Reconstruction of Infectious Bronchitis Virus (IBV) QX Genotype (GI-19 Lineage) Reveals Different Population Dynamics and Spreading Patterns When Evaluated on Different Epidemiological Scales. PLoS ONE 2017, 12, e0184401. [Google Scholar] [CrossRef]
- Segalés, J.; Kekarainen, T.; Cortey, M. The Natural History of Porcine Circovirus Type 2: From an Inoffensive Virus to a Devastating Swine Disease? Vet. Microbiol. 2013, 165, 13–20. [Google Scholar] [CrossRef]
- Rux, J.J.; Kuser, P.R.; Burnett, R.M. Structural and Phylogenetic Analysis of Adenovirus Hexons by Use of High-Resolution X-Ray Crystallographic, Molecular Modeling, and Sequence-Based Methods. J. Virol. 2003, 77, 9553–9566. [Google Scholar] [CrossRef] [PubMed]
- Saban, S.D.; Silvestry, M.; Nemerow, G.R.; Stewart, P.L. Visualization of α-Helices in a 6-Ångstrom Resolution Cryoelectron Microscopy Structure of Adenovirus Allows Refinement of Capsid Protein Assignments. J. Virol. 2006, 80, 12049–12059. [Google Scholar] [CrossRef] [PubMed]
- Mo, J. Historical Investigation of Fowl Adenovirus Outbreaks in South Korea from 2007 to 2021: A Comprehensive Review. Viruses 2021, 13, 2256. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Legnardi, M.; Mescolini, G.; Tucciarone, C.M.; Lupini, C.; Quaglia, G.; Catelli, E.; Cecchinato, M. Avian Metapneumovirus Subtype B around Europe: A Phylodynamic Reconstruction. Vet. Res. 2020, 51, 88. [Google Scholar] [CrossRef]
- Franzo, G.; Faustini, G.; Legnardi, M.; Cecchinato, M.; Drigo, M.; Tucciarone, C.M. Phylodynamic and Phylogeographic Reconstruction of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) in Europe: Patterns and Determinants. Transbound. Emerg. Dis. 2022, 69, E2175–E2184. [Google Scholar] [CrossRef]
- Houta, M.H.; Hassan, K.E.; Legnardi, M.; Tucciarone, C.M.; Abdel-Moneim, A.S.; Cecchinato, M.; El-Sawah, A.A.; Ali, A.; Franzo, G. Phylodynamic and Recombination Analyses of Avian Infectious Bronchitis Gi-23 Reveal a Widespread Recombinant Cluster and New among-Countries Linkages. Animals 2021, 11, 3182. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzo, G.; Faustini, G.; Tucciarone, C.M.; Pasotto, D.; Legnardi, M.; Cecchinato, M. Conflicting Evidence between Clinical Perception and Molecular Epidemiology: The Case of Fowl Adenovirus D. Animals 2023, 13, 3851. https://doi.org/10.3390/ani13243851
Franzo G, Faustini G, Tucciarone CM, Pasotto D, Legnardi M, Cecchinato M. Conflicting Evidence between Clinical Perception and Molecular Epidemiology: The Case of Fowl Adenovirus D. Animals. 2023; 13(24):3851. https://doi.org/10.3390/ani13243851
Chicago/Turabian StyleFranzo, Giovanni, Giulia Faustini, Claudia Maria Tucciarone, Daniela Pasotto, Matteo Legnardi, and Mattia Cecchinato. 2023. "Conflicting Evidence between Clinical Perception and Molecular Epidemiology: The Case of Fowl Adenovirus D" Animals 13, no. 24: 3851. https://doi.org/10.3390/ani13243851