
Comparison of Fecal Microbiota Communities between Primiparous and Multiparous Cows during Non-Pregnancy and Pregnancy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. DNA Extraction, PCR Amplification and Gene Sequencing
2.4. Data Analysis
3. Results
3.1. Sequencing Information
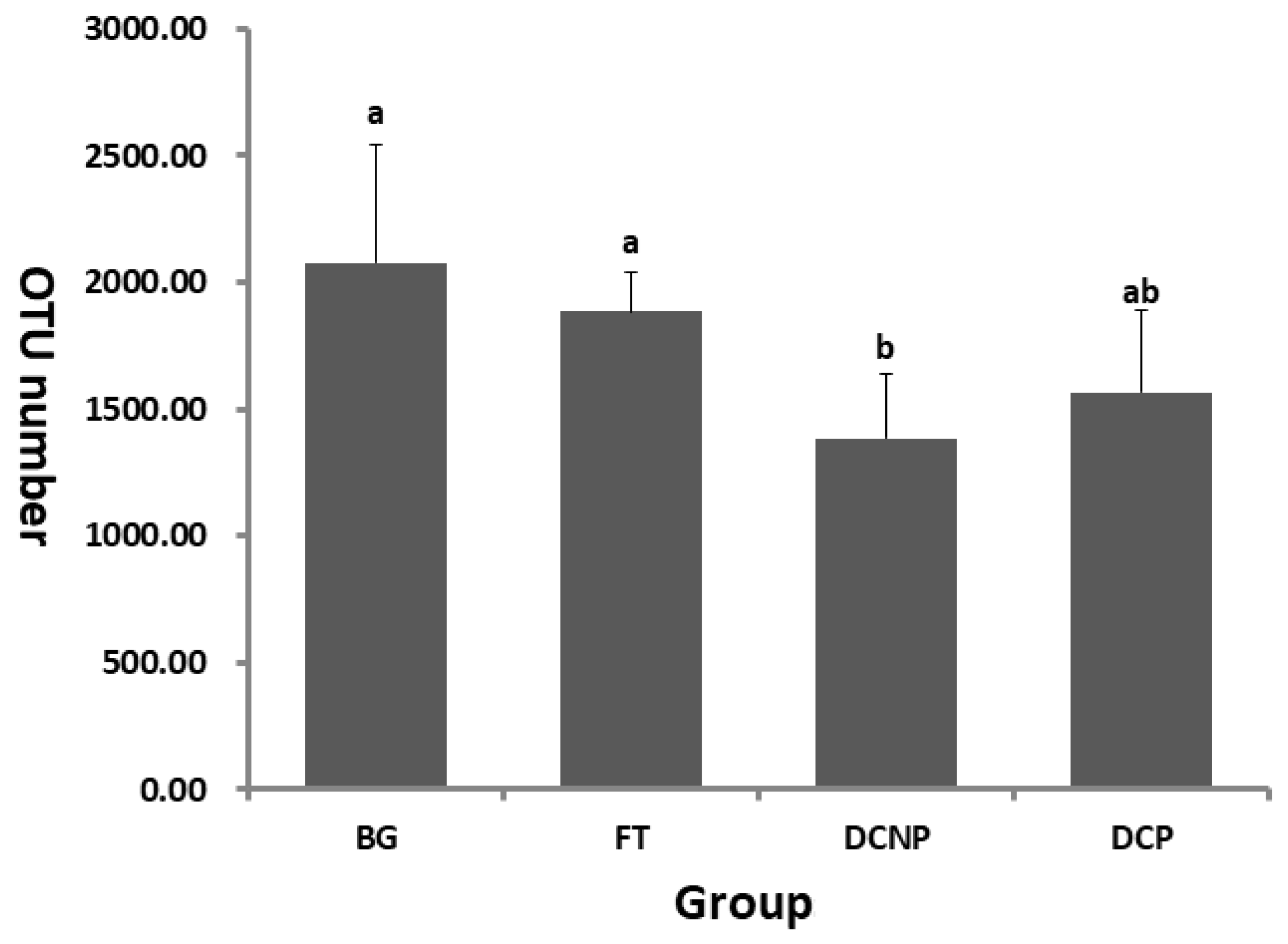
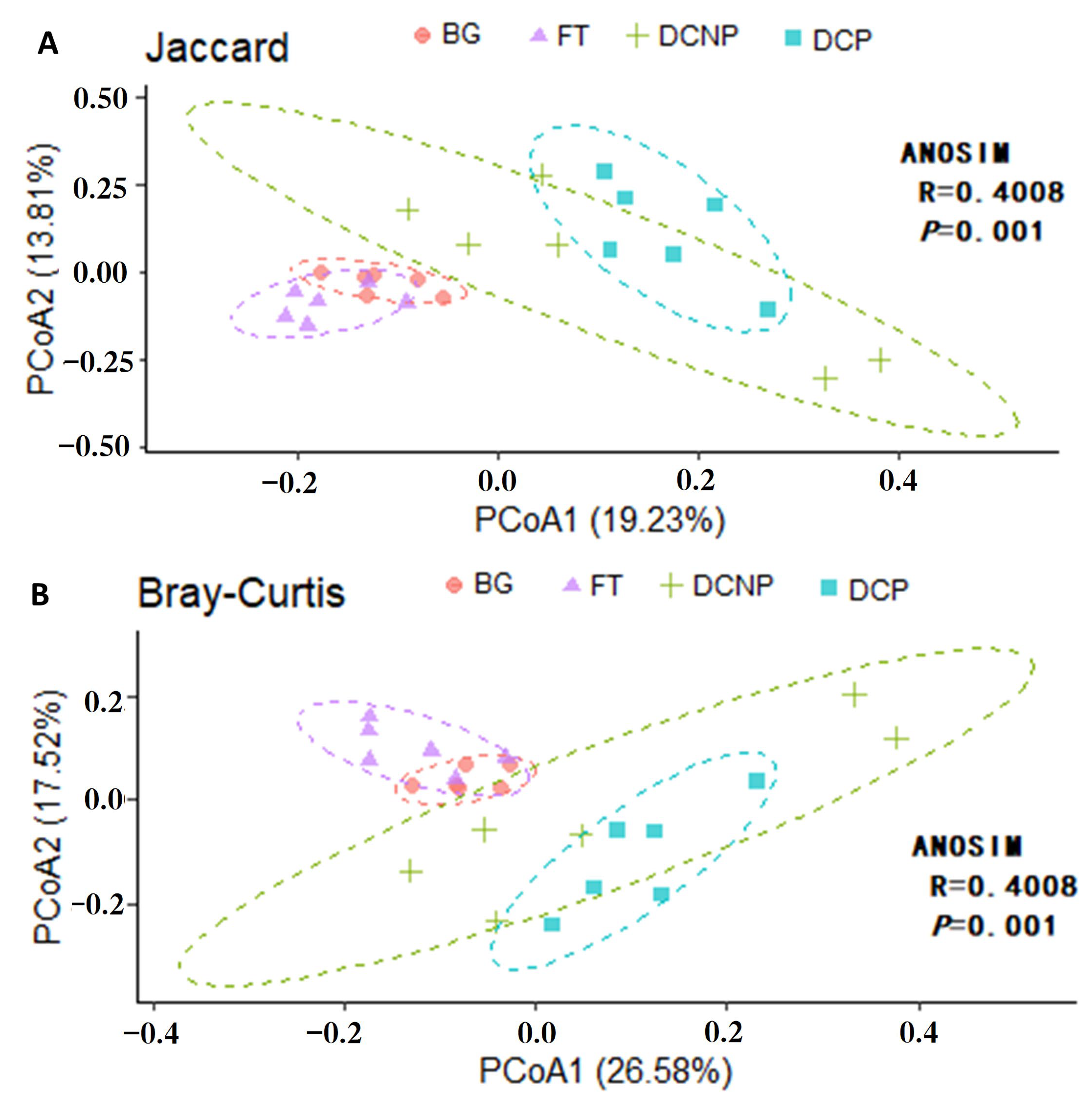
3.2. Microbial Ecology of the Fecal Microbiome
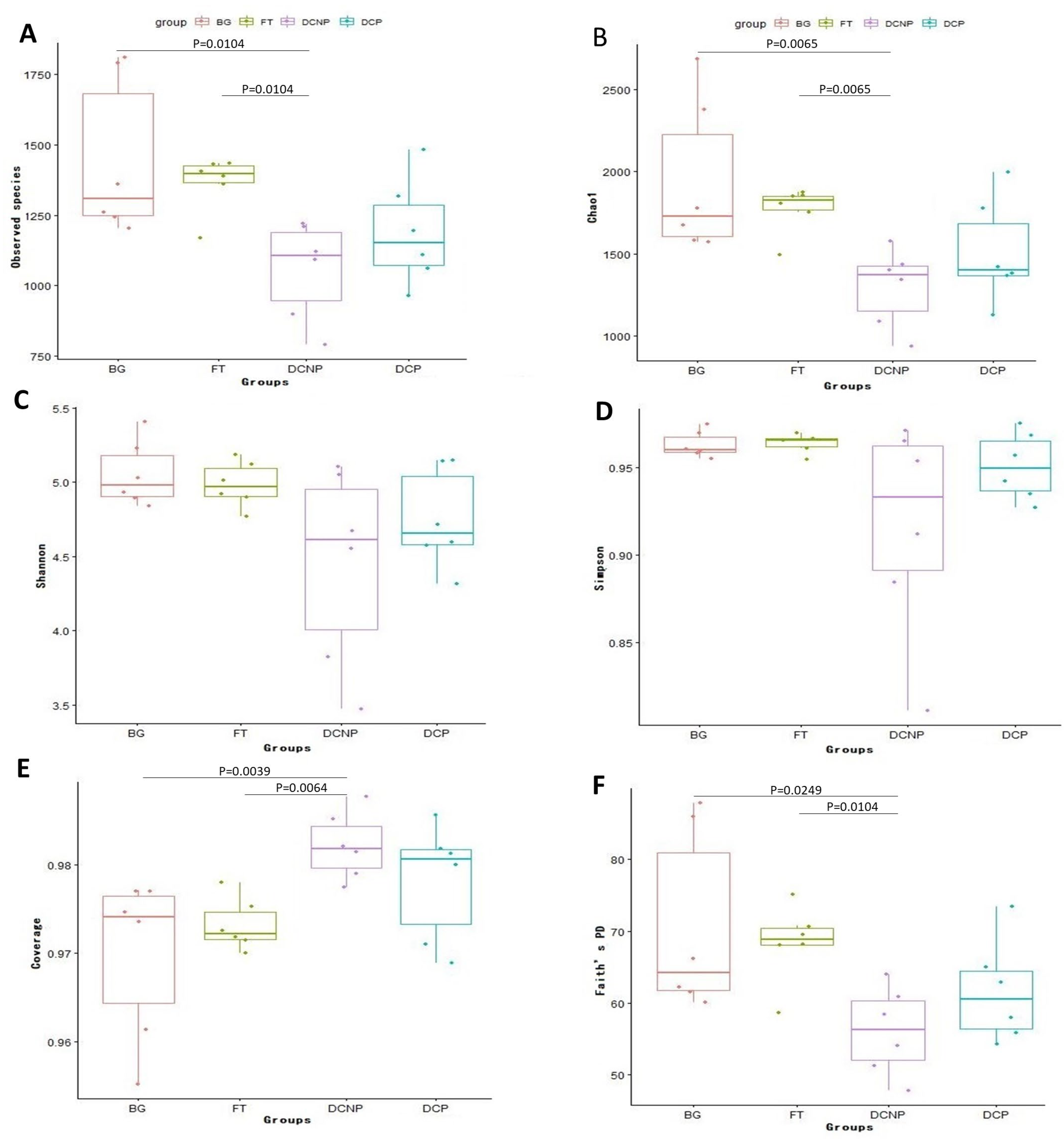
3.3. Microbial Diversity of the Fecal Microbiome
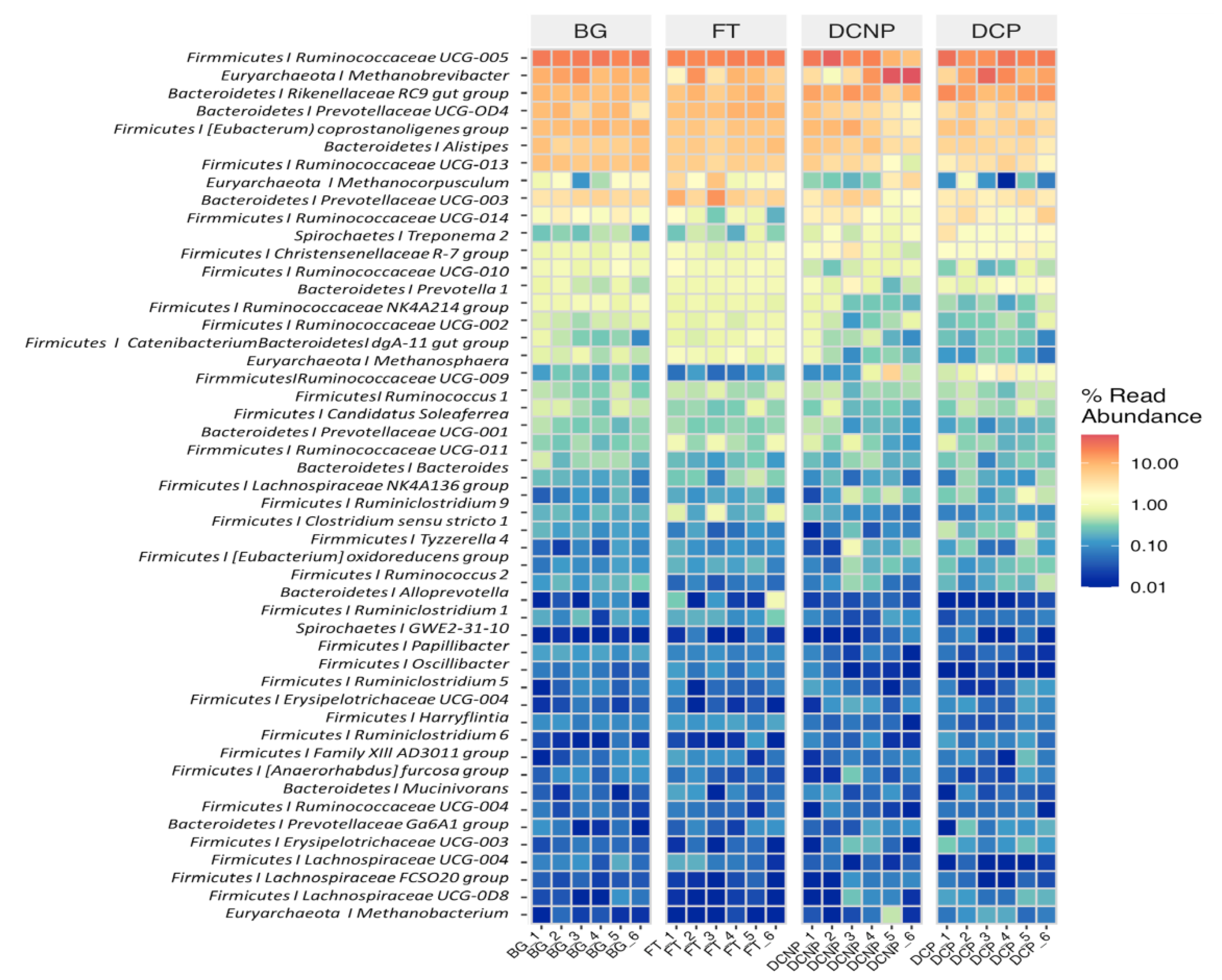
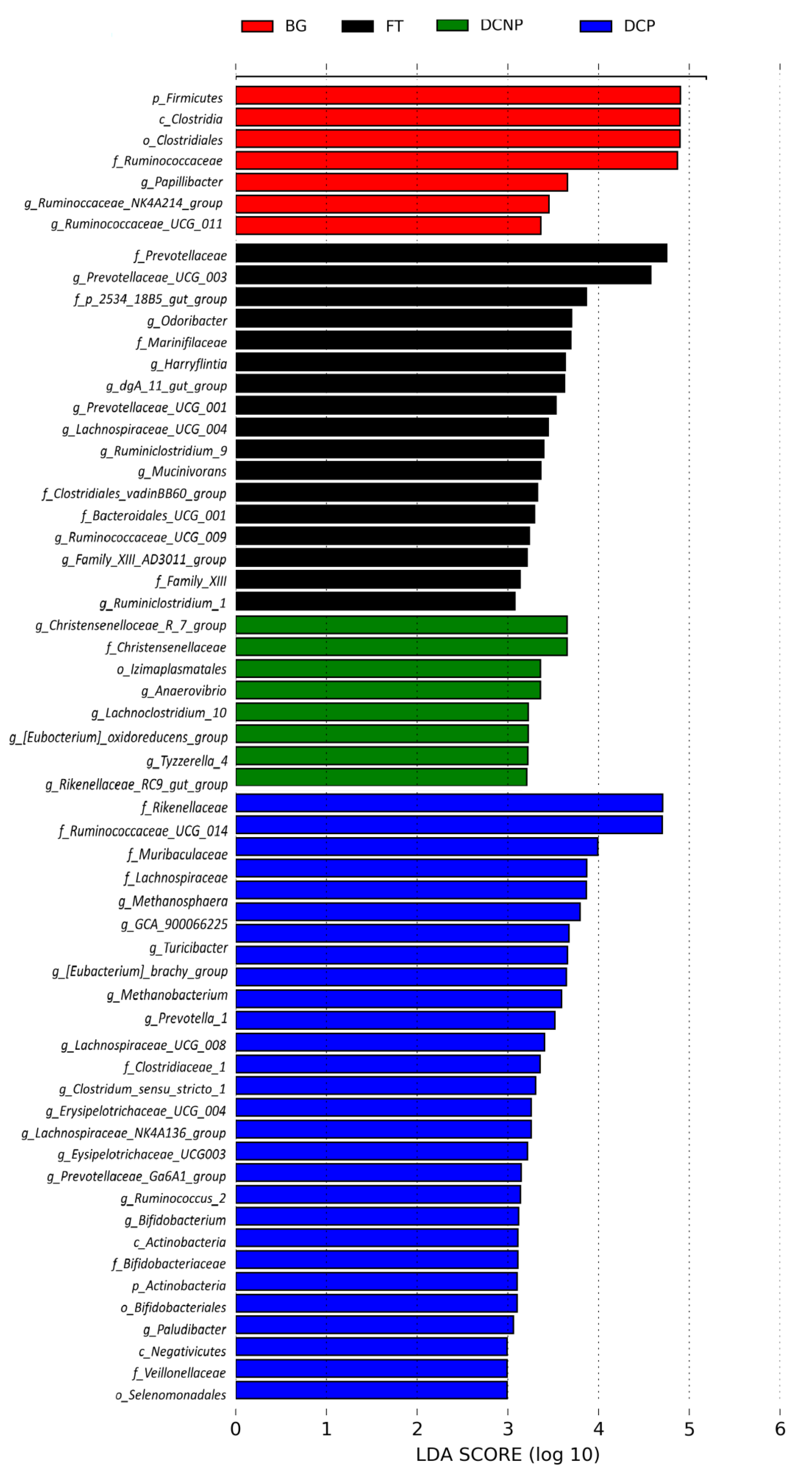
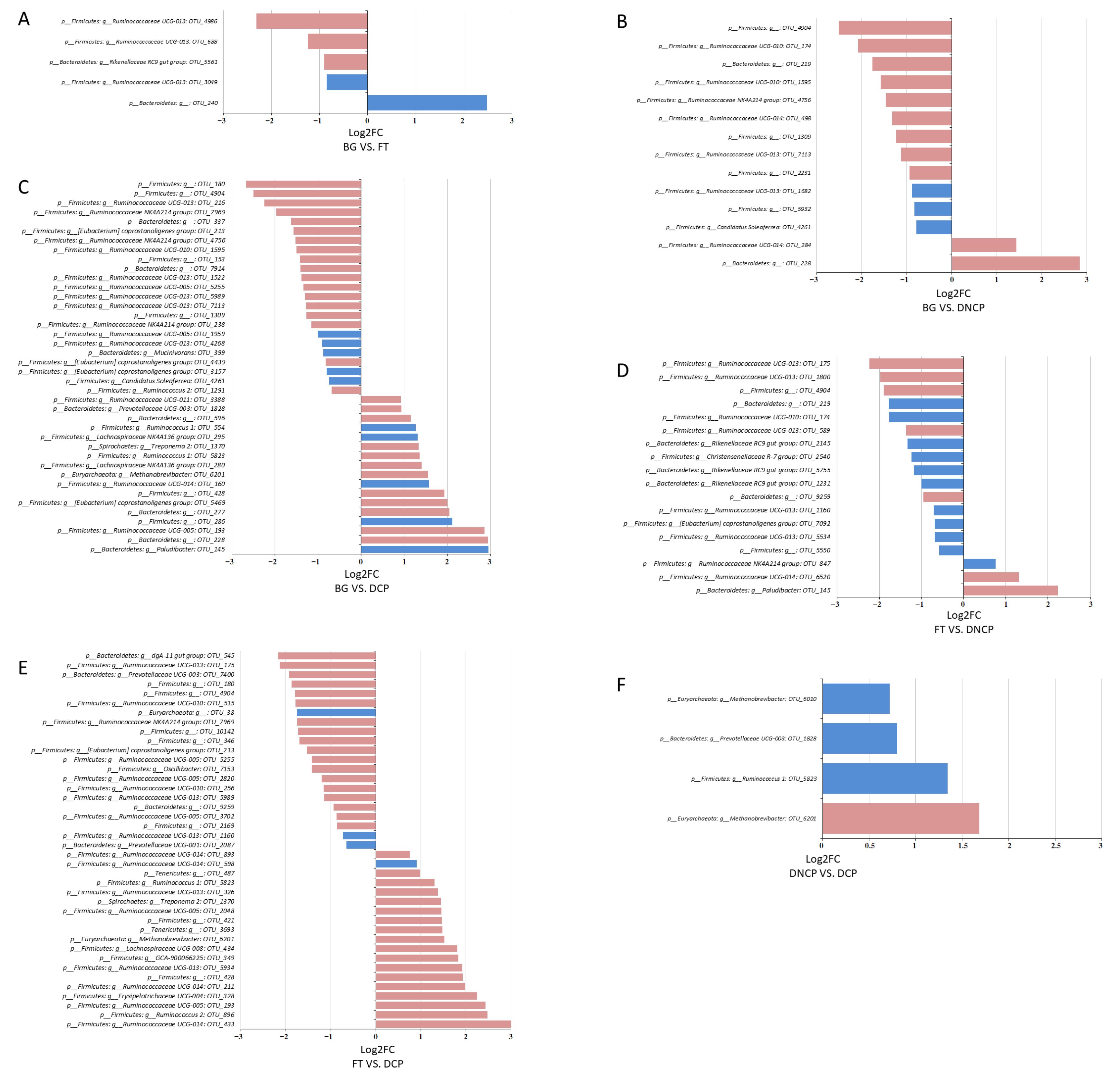
3.4. Microbial Taxonomy and Function Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Masko, M.; Zdrojkowski, L.; Wierzbicka, M.; Domino, M. Association between the Area of the Highest Flank Temperature and Concentrations of Reproductive Hormones during Pregnancy in Polish Konik Horses-A Preliminary Study. Animals 2021, 11, 1517. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, U.; Knorr, S.; Fuglsang, J.; Ovesen, P. Determinants of Maternal Insulin Resistance during Pregnancy: An Updated Overview. J. Diabetes Res. 2019, 2019, 5320156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, T.L. Symposium review: Immunological detection of the bovine conceptus during early pregnancy. J. Dairy Sci. 2019, 102, 3766–3777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieffer, T.E.; Faas, M.M.; Scherjon, S.A.; Prins, J.R. Pregnancy persistently affects memory T cell populations. J. Reprod. Immunol. 2017, 119, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradmark, A.; Pomeroy, J.; Renstrom, F.; Steiginga, S.; Persson, M.; Wright, A.; Bluck, L.; Domellof, M.; Kahn, S.E.; Mogren, I.; et al. Physical activity, sedentary behaviors, and estimated insulin sensitivity and secretion in pregnant and non-pregnant women. BMC Pregnancy Childb. 2011, 11, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovat, N.E.J.; Legare, D.J.; Gieni, R.S.; Lautt, W.W. Gestational postprandial insulin sensitivity in the Sprague Dawley rat: The putative role of hepatic insulin sensitizing substance in glucose partitioning in pregnancy. Can. J. Physiol. Pharmacol. 2020, 98, 541–547. [Google Scholar] [CrossRef] [PubMed]
- To, W.W.; Wong, M.W. Body fat composition and weight changes during pregnancy and 6-8 months post-partum in primiparous and multiparous women. Aust. N. Z. J. Obstet. Gynaecol. 2009, 49, 34–38. [Google Scholar] [CrossRef]
- Janovick, N.A.; Drackley, J.K. Prepartum dietary management of energy intake affects postpartum intake and lactation performance by primiparous and multiparous Holstein cows. J. Dairy Sci. 2010, 93, 3086–3102. [Google Scholar] [CrossRef] [PubMed]
- Janovick, N.A.; Boisclair, Y.R.; Drackley, J.K. Prepartum dietary energy intake affects metabolism and health during the periparturient period in primiparous and multiparous Holstein cows. J. Dairy Sci. 2011, 94, 1385–1400. [Google Scholar] [CrossRef]
- Chen, P.; Xu, T.T.; Zhang, C.D.; Tong, X.S.; Shaukat, A.; He, Y.F.; Liu, K.L.; Huang, S.C. Effects of Probiotics and Gut Microbiota on Bone Metabolism in Chickens: A Review. Metabolites 2022, 12, 1000. [Google Scholar] [CrossRef]
- Huang, S.C.; Zhang, C.D.; Xu, T.T.; Shaukat, A.; He, Y.F.; Chen, P.; Lin, L.X.; Yue, K.; Cao, Q.Q.; Tong, X.S. Integrated Fecal Microbiome and Metabolomics Reveals a Novel Potential Biomarker for Predicting Tibial Dyschondroplasia in Chickens. Front. Physiol. 2022, 13, 892. [Google Scholar] [CrossRef] [PubMed]
- Welch, C.B.; Ryman, V.E.; Pringle, T.D.; Lourenco, J.M. Utilizing the Gastrointestinal Microbiota to Modulate Cattle Health through the Microbiome-Gut-Organ Axes. Microorganisms 2022, 10, 1391. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, X.; Atwill, E.R.; Aly, S.S.; Williams, D.R.; Su, Z. Dynamic changes in fecal bacterial microbiota of dairy cattle across the production line. BMC Microbiol. 2022, 22, 132. [Google Scholar] [CrossRef] [PubMed]
- Gohir, W.; Whelan, F.J.; Surette, M.G.; Moore, C.; Schertzer, J.D.; Sloboda, D.M. Pregnancy-related changes in the maternal gut microbiota are dependent upon the mother’s periconceptional diet. Gut Microbes 2015, 6, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Backhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Liu, Y.; Wang, S.; Ma, J.; Yang, H. Distribution characteristics of intestinal microbiota during pregnancy and postpartum in healthy women. J. Matern. Fetal Neonatal Med. 2022, 35, 2915–2922. [Google Scholar] [CrossRef]
- Wang, J.; Shi, Z.H.; Yang, J.; Wei, Y.; Wang, X.Y.; Zhao, Y.Y. Gut microbiota dysbiosis in preeclampsia patients in the second and third trimesters. Chin. Med. J.-Peking 2020, 133, 1057–1065. [Google Scholar] [CrossRef]
- Crusell, M.K.W.; Hansen, T.H.; Nielsen, T.; Allin, K.H.; Ruhlemann, M.C.; Damm, P.; Vestergaard, H.; Rorbye, C.; Jorgensen, N.R.; Christiansen, O.B.; et al. Gestational diabetes is associated with change in the gut microbiota composition in third trimester of pregnancy and postpartum. Microbiome 2018, 6, 89. [Google Scholar] [CrossRef]
- Berry, A.S.F.; Pierdon, M.K.; Misic, A.M.; Sullivan, M.C.; O’Brien, K.; Chen, Y.; Murray, S.J.; Ramharack, L.A.; Baldassano, R.N.; Parsons, T.D.; et al. Remodeling of the maternal gut microbiome during pregnancy is shaped by parity. Microbiome 2021, 9, 146. [Google Scholar] [CrossRef]
- Gaukroger, C.H.; Edwards, S.A.; Walshaw, J.; Nelson, A.; Adams, I.P.; Stewart, C.J.; Kyriazakis, I. Shifting sows: Longitudinal changes in the periparturient faecal microbiota of primiparous and multiparous sows. Animal 2021, 15, 100135. [Google Scholar] [CrossRef]
- Pitta, D.W.; Indugu, N.; Kumar, S.; Vecchiarelli, B.; Sinha, R.; Baker, L.D.; Bhukya, B.; Ferguson, J.D. Metagenomic assessment of the functional potential of the rumen microbiome in Holstein dairy cows. Anaerobe 2016, 38, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Bogado Pascottini, O.; Spricigo, J.F.W.; Van Schyndel, S.J.; Mion, B.; Rousseau, J.; Weese, J.S.; LeBlanc, S.J. Effects of parity, blood progesterone, and non-steroidal anti-inflammatory treatment on the dynamics of the uterine microbiota of healthy postpartum dairy cows. PLoS ONE 2021, 16, e0233943. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Kapourchali, F.R.; Cresci, G.A.M. Early-Life Gut Microbiome-The Importance of Maternal and Infant Factors in Its Establishment. Nutr. Clin. Pract. 2020, 35, 386–405. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, S.M.; Cunningham, S.A.; Dunlop, A.L.; Corwin, E.J. The Maternal Gut Microbiome During Pregnancy. MCN Am. J. Matern. Child Nurs. 2017, 42, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Smid, M.C.; Ricks, N.M.; Panzer, A.; McCoy, A.N.; Azcarate-Peril, M.A.; Keku, T.O.; Boggess, K.A. Maternal Gut Microbiome Biodiversity in Pregnancy. Am. J. Perinatol. 2018, 35, 24–30. [Google Scholar] [PubMed]
- Williams, C.L.; Garcia-Reyero, N.; Martyniuk, C.J.; Tubbs, C.W.; Bisesi, J.H., Jr. Regulation of endocrine systems by the microbiome: Perspectives from comparative animal models. Gen. Comp. Endocrinol. 2020, 292, 113437. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Adnane, M.; Archunan, G. Significance of cervico-vaginal microbes in bovine reproduction and pheromone production—A hypothetical review. Res. Vet. Sci. 2021, 135, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Deng, F.; Zhang, M.; Jia, X.; Lai, S.J. Characterization of Vaginal Microbiota Associated with Pregnancy Outcomes of Artificial Insemination in Dairy Cows. J. Microbiol. Biotechnol. 2020, 30, 804–810. [Google Scholar] [CrossRef]
- Deng, F.L.; McClure, M.; Rorie, R.; Wang, X.F.; Chai, J.M.; Wei, X.Y.; Lai, S.J.; Zhao, J.C. The vaginal and fecal microbiomes are related to pregnancy status in beef heifers. J. Anim. Sci. Biotechnol. 2019, 10, 92. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Wells, J.E. A Meta-analysis of Bacterial Diversity in the Feces of Cattle. Curr. Microbiol. 2016, 72, 145–151. [Google Scholar] [CrossRef]
- Hagey, J.V.; Bhatnagar, S.; Heguy, J.M.; Karle, B.M.; Price, P.L.; Meyer, D.; Maga, E.A. Fecal Microbial Communities in a Large Representative Cohort of California Dairy Cows. Front. Microbiol. 2019, 10, 1093. [Google Scholar] [CrossRef]
- Uchiyama, J.; Murakami, H.; Sato, R.; Mizukami, K.; Suzuki, T.; Shima, A.; Ishihara, G.; Sogawa, K.; Sakaguchi, M. Examination of the fecal microbiota in dairy cows infected with bovine leukemia virus. Vet. Microbiol. 2020, 240, 108547. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Lin, X.; Wang, Z.; Hou, Q.; Wang, Y.; Hu, Z. High-production dairy cattle exhibit different rumen and fecal bacterial community and rumen metabolite profile than low-production cattle. MicrobiologyOpen 2019, 8, e00673. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Kang, K.; Wang, H.; Wang, Z.; Xue, B.; Wang, L.; Xu, F.; Peng, Q. Effects of dietary supplementation of active dried yeast on fecal methanogenic archaea diversity in dairy cows. Anaerobe 2017, 44, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Miranda-CasoLuengo, R.; Lu, J.N.; Williams, E.J.; Miranda-CasoLuengo, A.A.; Carrington, S.D.; Evans, A.C.O.; Meijer, W.G. Delayed differentiation of vaginal and uterine microbiomes in dairy cows developing postpartum endometritis. PLoS ONE 2019, 14, e0200974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ault, T.B.; Clemmons, B.A.; Reese, S.T.; Dantas, F.G.; Franco, G.A.; Smith, T.P.L.; Edwards, J.L.; Myer, P.R.; Pohler, K.G. Uterine and vaginal bacterial community diversity prior to artificial insemination between pregnant and nonpregnant postpartum cows. J. Anim. Sci. 2019, 97, 4298–4304. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.T.; Kong, Q.M.; Li, X.; Zhao, J.X.; Zhang, H.; Chen, W.; Wang, G. A High-Fat Diet Increases Gut Microbiota Biodiversity and Energy Expenditure due to Nutrient Difference. Nutrients 2020, 12, 3197. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, J.A.; Peters, S.O.; De Donato, M.; Cervantes, A.P.; Ogunade, I.M. Effects of a blend of Saccharomyces cerevisiae-based direct-fed microbial and fermentation products on plasma carbonyl-metabolome and fecal bacterial community of beef steers. J. Anim. Sci. Biotechnol. 2020, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, G.O.; Gruninger, R.J.; Jones, D.R.; Beauchemin, K.A.; Yang, W.Z.; Wang, Y.; Abbott, D.W.; Tsang, A.; McAllister, T.A. Effect of ammonia fiber expansion-treated wheat straw and a recombinant fibrolytic enzyme on rumen microbiota and fermentation parameters, total tract digestibility, and performance of lambs. J. Anim. Sci. 2020, 98, skaa116. [Google Scholar] [CrossRef]
- Ren, H.; Su, X.; Bai, H.; Yang, Y.; Wang, H.; Dan, Z.; Lu, J.; Wu, S.; Cai, C.; Cao, Y.; et al. Specific enrichment of microbes and increased ruminal propionate production: The potential mechanism underlying the high energy efficiency of Holstein heifers fed steam-flaked corn. AMB Express 2019, 9, 209. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Huang, S.; Si, J.; Zhang, J.; Gaowa, N.; Sun, X.; Lv, J.; Liu, G.; He, Y.; Wang, W.; et al. Effects of Paper Mulberry Silage on the Milk Production, Apparent Digestibility, Antioxidant Capacity, and Fecal Bacteria Composition in Holstein Dairy Cows. Animals 2020, 10, 1152. [Google Scholar] [CrossRef]
- Paul, S.S.; Dey, A.; Baro, D.; Punia, B.S. Comparative community structure of archaea in rumen of buffaloes and cattle. J. Sci. Food Agric. 2017, 97, 3284–3293. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Miller, T.L. Phylogenetic analysis of Methanobrevibacter isolated from feces of humans and other animals. Arch. Microbiol. 1998, 169, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Batt, S.M.; Wannemuehler, M.; Dispirito, A.; Beitz, D.C. Effect of feeding of a cholesterol-reducing bacterium, Eubacterium coprostanoligenes, to germ-free mice. Lab. Anim. Sci. 1998, 48, 253–255. [Google Scholar] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, X.; He, Y.; Kang, Z.; Chen, S.; Sun, W.; Wang, J.; Lai, S. Comparison of Fecal Microbiota Communities between Primiparous and Multiparous Cows during Non-Pregnancy and Pregnancy. Animals 2023, 13, 869. https://doi.org/10.3390/ani13050869
Jia X, He Y, Kang Z, Chen S, Sun W, Wang J, Lai S. Comparison of Fecal Microbiota Communities between Primiparous and Multiparous Cows during Non-Pregnancy and Pregnancy. Animals. 2023; 13(5):869. https://doi.org/10.3390/ani13050869
Chicago/Turabian StyleJia, Xianbo, Yang He, Zhe Kang, Shiyi Chen, Wenqiang Sun, Jie Wang, and Songjia Lai. 2023. "Comparison of Fecal Microbiota Communities between Primiparous and Multiparous Cows during Non-Pregnancy and Pregnancy" Animals 13, no. 5: 869. https://doi.org/10.3390/ani13050869