Fermented Total Mixed Ration Alters Rumen Fermentation Parameters and Microbiota in Dairy Cows

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Experimental, and Design Diets
2.2. Rumen Sample Collection and Measurements
2.3. Microbial DNA Extraction, PCR Amplification, and Illumina MiSeq Sequencing
2.4. Sequencing Data Processing
2.5. Statistical Analyses
3. Results
3.1. Rumen Fermentation Parameter
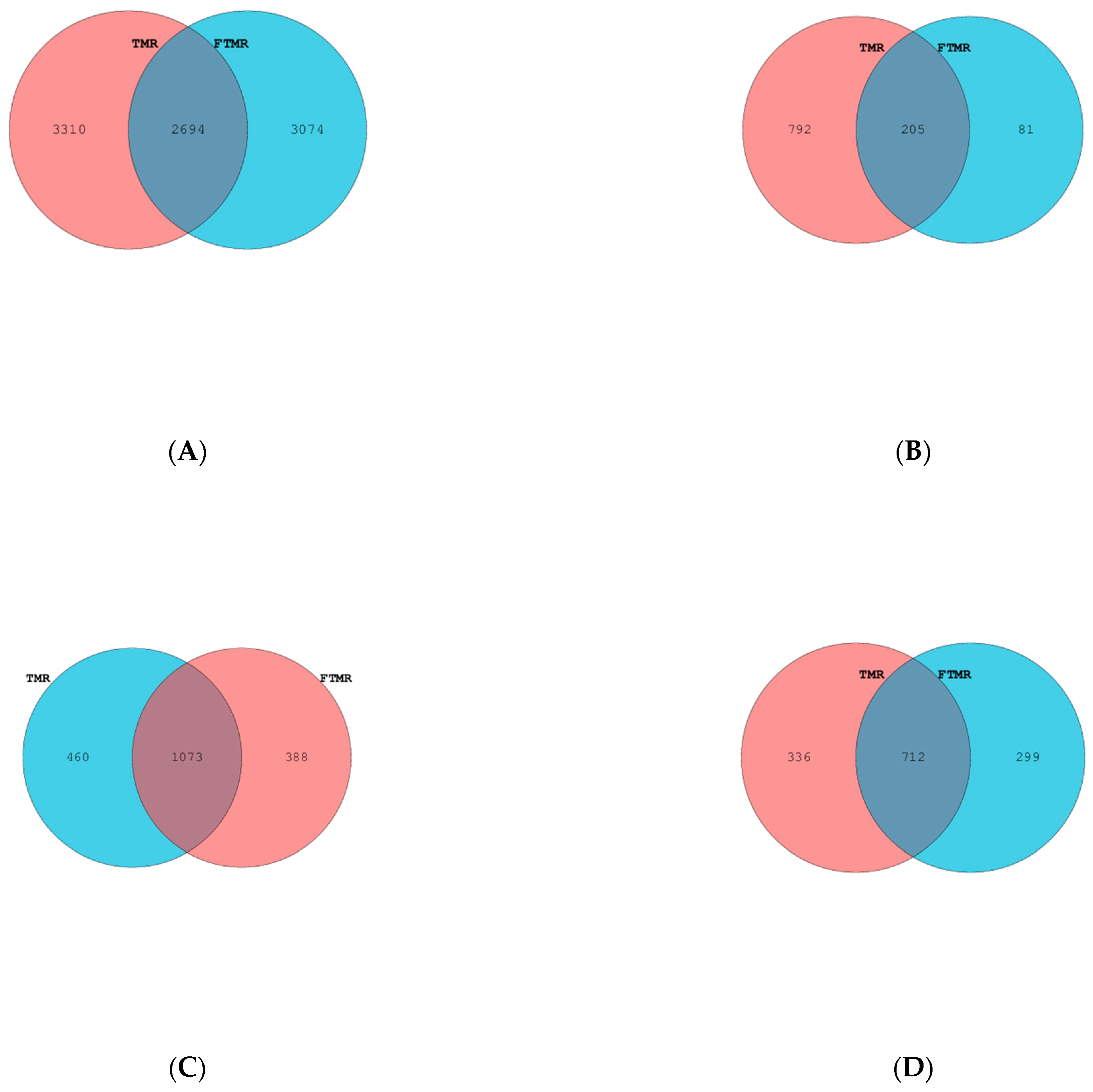
3.2. Sequence, Diversity, Species Richness, and Composition of Bacterial Microbiota
3.3. Sequence, Diversity, Species Richness, and Composition of Anaerobic Fungal Microbiota
3.4. Sequence, Diversity, Species Richness, and Composition of Ciliate Protozoal Microbiota
3.5. Sequence, Diversity, Species Richness, and Composition of Archaeal Microbiota
3.6. The Correlations between the Relative Abundance of Predominant Ruminal Microbiota and Rumen Fermentation Parameter
3.7. Prediction of Different Treatment Functions of Rumen Bacteria
4. Discussion
4.1. Rumen Fermentation Parameter
4.2. Diversity, Species Richness, and Composition of Bacterial, Anaerobic Fungal, Ciliate Protozoal, and Archaeal Microbiota
4.3. Correlations between the Relative Abundance of Predominant Ruminal Microbiota and Rumen Fermentation Parameter
4.4. Function Prediction of Rumen Bacteria
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hobson, P.N.; Stewart, C.S. The Rumen Microbial Ecosystem; Springer: Berlin/Heidelberg, Germany, 1997. [Google Scholar]
- Shabat, S.K.B.; Sasson, G.; Doronfaigenboim, A.; Durman, T.; Yaacoby, S.; Miller, M.E.B.; White, B.A.; Shterzer, N.; Mizrahi, I. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Peng, Y.J.; Chen, Y.; Klinger, C.M.; Oba, M.; Liu, J.X.; Guan, L.L. Assessment of microbiome changes after rumen transfaunation: Implications on improving feed efficiency in beef cattle. Microbiome 2018, 6, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Guan, L.L. Metatranscriptomic Profiling Reveals Linkages between the Active Rumen Microbiome and Feed Efficiency in Beef Cattle. Appl. Environ. Microbiol. 2017, 83, e00061-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Indugu, N.; Vecchiarelli, B.; Pitta, D.W. Associative patterns among anaerobic fungi, methanogenic archaea, and bacterial communities in response to changes in diet and age in the rumen of dairy cows. Front. Microbiol. 2015, 6, 781. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Takahashi, T.; Horiguchi, K. Effects of addition of food by-products on the fermentation quality of a total mixed ration with whole crop rice and its digestibility, preference, and rumen fermentation in sheep. Anim. Feed. Sci. Technol. 2009, 151, 1–11. [Google Scholar]
- Wang, F.; Nishino, N. Ensiling of soybean curd residue and wet brewers grains with or without other feeds as a total mixed ration. J. Dairy Sci. 2008, 91, 2380–2387. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, J.; Bu, D.; Guo, W.; Song, Z.; Zhang, J. Effect of storing total mixed rations anaerobically in bales on feed quality. Anim. Feed Sci. Technol. 2010, 161, 94–102. [Google Scholar] [CrossRef]
- Han, H.; Ogata, Y.; Yamamoto, Y.; Nagao, S.; Nishino, N. Identification of lactic acid bacteria in the rumen and feces of dairy cows fed total mixed ration silage to assess the survival of silage bacteria in the gut. J. Dairy Sci. 2014, 97, 5754–5762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Guo, G.; Wen, A.; Desta, S.T.; Wang, J.; Wang, Y.; Shao, T. The effect of different additives on the fermentation quality, in vitro digestibility and aerobic stability of a total mixed ration silage. Anim. Feed Sci. Technol. 2015, 207, 41–50. [Google Scholar] [CrossRef]
- Wang, C.; Nishino, N. Presence of sourdough lactic acid bacteria in commercial total mixed ration silage as revealed by denaturing gradient gel electrophoresis analysis. Lett. Appl. Microbiol. 2010, 51, 436–442. [Google Scholar] [CrossRef]
- Liu, Q.H.; Xiang-Yu, L.I.; Desta, S.T.; Zhang, J.G.; Shao, T. Effects of Lactobacillus plantarum and fibrolytic enzyme on the fermentation quality and in vitro digestibility of total mixed rations silage including rape straw. J. Integr. Agric. 2016, 15, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Nishino, N.; Wada, H.; Yoshida, M.; Shiota, H. Microbial counts, fermentation products, and aerobic stability of whole crop corn and a total mixed ration ensiled with and without inoculation of Lactobacillus casei or Lactobacillus buchneri. J. Dairy Sci. 2004, 87, 2563–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li Ying, H.; Borjigin, N.; Yu, Z. Effect of inoculants and fibrolytic enzymes on the fermentation characteristics, in vitro digestibility and aflatoxins accumulation of whole-crop corn silage. Grassl. Sci. 2017, 63, 69–78. [Google Scholar] [CrossRef]
- Cao, Y.; Takahashi, T.; Horiguchi, K.-i.; Yoshida, N.; Cai, Y. Methane emissions from sheep fed fermented or non-fermented total mixed ration containing whole-crop rice and rice bran. Anim. Feed Sci. Technol. 2010, 157, 72–78. [Google Scholar] [CrossRef]
- Cao, Y.; Zang, Y.; Jiang, Z.; Han, Y.; Hou, J.J.; Liu, H.; Zhong, R.; Fang, J.; Zhang, A.; Yoshida, N. Fermentation quality and nutritive value of fresh and fermented total mixed rations containing Chinese wildrye or corn stover. Grassl. Sci. 2016, 62, 213–223. [Google Scholar] [CrossRef]
- Kung, L.; Muck, R. Animal response to silage additives. In Proceedings of the Conference on Silage: Field to Feedbunk, Madison, WI, USA, 13 February 1997. [Google Scholar]
- Kung, L. Potential Factors that May Limit the Effectiveness of Silage Additives. In Proceedings of the XV International Silage Conference, Madison, WI, USA, 27 July 2009. [Google Scholar]
- Bryant, M.P.; Burkey, L.A. Numbers and some predominant groups of bacteria in the rumen of cows fed different rations. J. Dairy Sci. 1953, 36, 218–224. [Google Scholar] [CrossRef]
- Warner, A.C.I. Some Factors Influencing the Rumen Microbial Population. J. Gen. Microbiol. 1962, 28, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Leedle, J.A.; Bryant, M.P.; Hespell, R.B. Diurnal variations in bacterial numbers and fluid parameters in ruminal contents of animals fed low- or high-forage diets. Appl. Environ. Microbiol. 1982, 44, 402–412. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, Z.G.; Muck, R.E.; Weimer, P.J. The survival of silage inoculant lactic acid bacteria in rumen fluid. J. Appl. Microbiol. 2010, 94, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, R.; Stevenson, D.; Beauchemin, K.; Muck, R.; Weimer, P. Changes in ruminal bacterial community composition following feeding of alfalfa ensiled with a lactic acid bacterial inoculant. J. Dairy Sci. 2012, 95, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Govea, F.E.; Muck, R.E.; Mertens, D.R.; Weimer, P.J. Microbial inoculant effects on silage and in vitro ruminal fermentation, and microbial biomass estimation for alfalfa, bmr corn, and corn silages. Anim. Feed Sci. Technol. 2011, 163, 2–10. [Google Scholar] [CrossRef]
- Weimer, P.J. Redundancy, resilience, and host specificity of the ruminal microbiota: Implications for engineering improved ruminal fermentations. Front. Microbiol. 2014, 6, 296. [Google Scholar] [CrossRef] [Green Version]
- Malmuthuge, N.; Guan, L.L. Understanding host-microbial interactions in rumen: Searching the best opportunity for microbiota manipulation. J. Anim. Sci. Biotechnol. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boston, R.C.; Fox, D.G.; Sniffen, C.; Janczewski, E.; Munson, R.; Chalupa, W. The conversion of a scientific model describing dairy cow nutrition and production to an industry tool: The CPM Dairy project. In Modelling Nutrient Utilization in Farm Animals; CABI: Wallingford, UK, 2000; pp. 361–377. [Google Scholar]
- Wang, B.; Mao, S.Y.; Yang, H.J.; Wu, Y.M.; Wang, J.K.; Li, S.L.; Shen, Z.M.; Liu, J.X. Effects of alfalfa and cereal straw as a forage source on nutrient digestibility and lactation performance in lactating dairy cows. J. Dairy Sci. 2014, 97, 7706–7715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, X.; Gao, H.; Wang, X.; Zhang, G.; Zhang, Y. Replacing alfalfa hay with dry corn gluten feed and Chinese wild rye grass: Effects on rumen fermentation, rumen microbial protein synthesis, and lactation performance in lactating dairy cows. J. Dairy Sci. 2017, 100, 2672–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shi, H.; Wang, Y.; Li, S.; Cao, Z.; Ji, S.; He, Y.; Zhang, H. Effect of dietary forage to concentrate ratios on dynamic profile changes and interactions of ruminal microbiota and metabolites in Holstein heifers. Front. Microbiol. 2017, 8, 2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius, A.G.; Kittelmann, S.; Macdonald, K.A.; Waghorn, G.C.; Janssen, P.H.; Sikkema, E. Nitrogen metabolism and rumen microbial enumeration in lactating cows with divergent residual feed intake fed high-digestibility pasture. J. Dairy Sci. 2012, 95, 5024–5034. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460. [Google Scholar] [CrossRef] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.; Koetschan, C.; Müller, T. ITS2, 18S, 16S or any other RNA—Simply aligning sequences and their individual secondary structures simultaneously by an automatic approach. Gene 2014, 546, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; et al. UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, C.; Kristensen, N.B. Nitrogen recycling through the gut and the nitrogen economy of ruminants: An asynchronous symbiosis. J. Anim. Sci. 2008, 86, E293–E305. [Google Scholar] [CrossRef] [Green Version]
- Brito, A.F.; Broderick, G.A.; Reynal, S.M. Effects of Different Protein Supplements on Omasal Nutrient Flow and Microbial Protein Synthesis in Lactating Dairy Cows1. J. Dairy Sci. 2007, 90, 1828–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yu, Q.; Wang, X.; Song, J.; Lambo, M.T.; Huang, J.; He, P.; Li, Y.; Zhang, Y. Replacing alfalfa hay with industrial hemp ethanol extraction byproduct and Chinese wildrye hay: Effects on lactation performance, plasma metabolites, and bacterial communities in Holstein cows. Front. Vet. Sci. 2023, 10, 1061219. [Google Scholar] [CrossRef] [PubMed]
- Hristov, A.; Ropp, J. Effect of dietary carbohydrate composition and availability on utilization of ruminal ammonia nitrogen for milk protein synthesis in dairy cows. J. Dairy Sci. 2003, 86, 2416–2427. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gao, J.; Lv, J.; Lambo, M.T.; Wang, Y.; Wang, L.; Zhang, Y. Replacing soybean meal with high-oil pumpkin seed cake in the diet of lactating Holstein dairy cows modulated rumen bacteria and milk fatty acid profile. J. Dairy Sci. 2023, 106, 1803–1814. [Google Scholar] [CrossRef] [PubMed]
- Miyaji, M.; Inoue, H.; Kawaide, T.; Tohno, M.; Kamiya, Y.; Nonaka, K. Effects of conservation method and crushing method of rice grain on rumen fermentation and nutrient digestibility in steers. Anim. Feed Sci. Technol. 2017, 227, 75–83. [Google Scholar] [CrossRef]
- Russell, J.B.; Wallace, R.J. Energy-Yielding and Energy-Consuming Reactions; Springer Nature: Switzerland, 1997; pp. 246–282. [Google Scholar]
- Woodford, S.T.; Murphy, M.R. Effect of forage physical form on chewing activity, dry matter intake, and rumen function of dairy cows in early lactation. J. Dairy Sci. 1988, 71, 674–686. [Google Scholar] [CrossRef]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [Green Version]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Collaborators, G.R.C.; Abecia, L.; Angarita, E.; Aravena, P.; Arenas, G.N. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef] [Green Version]
- Lan, M.; Yang, B.; Hu, X.; Yang, L.; Liu, J.; Yu, Z.; Wang, J. Comparative Analysis of the Microbiota Between Sheep Rumen and Rabbit Cecum Provides New Insight Into Their Differential Methane Production. Front. Microbiol. 2018, 9, 575. [Google Scholar]
- Shen, H.; Lu, Z.; Chen, Z.; Wu, Y.; Shen, Z. Rapid Fermentable Substance Modulates Interactions between Ruminal Commensals and Toll-Like Receptors in Promotion of Immune Tolerance of Goat Rumen. Front. Microbiol. 2016, 7, 1812. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Muller, M.; de Vos, W. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420. [Google Scholar] [CrossRef]
- Elie, J.; Itzhak, M. Composition and similarity of bovine rumen microbiota across individual animals. PLoS ONE 2012, 7, e33306. [Google Scholar]
- Zhou, Z.; Fang, L.; Meng, Q.; Li, S.; Chai, S.; Liu, S.; Schonewille, J.T. Assessment of Ruminal Bacterial and Archaeal Community Structure in Yak (Bos grunniens). Front. Microbiol. 2017, 8, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, V.; Petri, R.; Humer, E.; Kröger, I.; Mann, E.; Reisinger, N.; Wagner, M.; Zebeli, Q. High-grain diets supplemented with phytogenic compounds or autolyzed yeast modulate ruminal bacterial community and fermentation in dry cows. J. Dairy Sci. 2018, 101, 2335–2349. [Google Scholar] [CrossRef]
- Mao, S.Y.; Huo, W.J.; Zhu, W.Y. Microbiome–metabolome analysis reveals unhealthy alterations in the composition and metabolism of ruminal microbiota with increasing dietary grain in a goat model. Environ. Microbiol. 2015, 18, 525. [Google Scholar] [CrossRef]
- Sandra, K.; Henning, S.; Walters, W.A.; Clemente, J.C.; Rob, K.; Gordon, J.I.; Janssen, P.H. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS ONE 2013, 8, e47879. [Google Scholar]
- Hook, S.E.; Steele, M.A.; Northwood, K.S.; Dijkstra, J.; France, J.; Wright, A.G.; Mcbride, B.W. Impact of subacute ruminal acidosis (SARA) adaptation and recovery on the density and diversity of bacteria in the rumen of dairy cows. FEMS Microbiol. Ecol. 2011, 78, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieho, K.; Bogert, B.V.D.; Henderson, G.; Bannink, A.; Ramiro-Garcia, J.; Smidt, H.; Dijkstra, J. Changes in rumen microbiota composition and in situ degradation kinetics during the dry period and early lactation as affected by rate of increase of concentrate allowance. J. Dairy Sci. 2017, 100, 2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goad, D.W.; Goad, C.L.; Nagaraja, T.G. Ruminal microbial and fermentative changes associated with experimentally induced subacute acidosis in steers. J. Anim. Sci. 1998, 76, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hristov, A.N.; Ivan, M.; Rode, L.M.; McAllister, T.A. Fermentation characteristics and ruminal ciliate protozoal populations in cattle fed medium- or high-concentrate barley-based diets. J. Anim. Sci. 2001, 79, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Danielsson, R.; Dicksved, J.; Sun, L.; Gonda, H.; Müller, B.; Schnürer, A.; Bertilsson, J. Methane Production in Dairy Cows Correlates with Rumen Methanogenic and Bacterial Community Structure. Front. Microbiol. 2017, 8, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leahy, S.C.; Kelly, W.J.; Ronimus, R.S.; Wedlock, N.; Altermann, E.; Attwood, G.T. Genome sequencing of rumen bacteria and archaea and its application to methane mitigation strategies. Animal 2013, 7, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Han, Y.; Ding, Y.; Zhu, B.; Song, S.; Xiao, H. Health effects of dietary sulfated polysaccharides from seafoods and their interaction with gut microbiota. Compr. Rev. Food Sci. Food Saf. 2021, 20, 2882–2913. [Google Scholar] [CrossRef]
- Bui, T.P.N.; Schols, H.A.; Jonathan, M.; Stams, A.J.M.; de Vos, W.M.; Plugge, C.M. Mutual Metabolic Interactions in Co-cultures of the Intestinal Anaerostipes rhamnosivorans With an Acetogen, Methanogen, or Pectin-Degrader Affecting Butyrate Production. Front. Microbiol. 2019, 10, 2449. [Google Scholar] [CrossRef] [Green Version]
- Clemmons, B.A.; Powers, J.B.; Campagna, S.R.; Seay, T.B.; Embree, M.M.; Myer, P.R. Rumen fluid metabolomics of beef steers differing in feed efficiency. Metabolomics 2020, 16, 23. [Google Scholar] [CrossRef]
- Arshad, U.; Zenobi, M.G.; Staples, C.R.; Santos, J.E.P. Meta-analysis of the effects of supplemental rumen-protected choline during the transition period on performance and health of parous dairy cows. J. Dairy Sci. 2020, 103, 282–300. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.E. Invited review: Improving feed efficiency in dairy production: Challenges and possibilities. Animal 2015, 9, 395–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Treatment | |
|---|---|---|
| TMR 1 | FTMR 1 | |
| Ingredient, % of DM | ||
| Alfalfa hay | 11.8 | 11.8 |
| Wet corn gluten feed | 6.9 | 6.9 |
| Corn stover | 3.4 | 3.4 |
| Corn silage | 27.8 | 27.8 |
| Ground corn | 24.6 | 24.6 |
| Soybean meal | 9.8 | 9.8 |
| Cottonseed meal | 4.9 | 4.9 |
| DDGS | 7.4 | 7.4 |
| Expanded soybean | 1.0 | 1.0 |
| Premix 2 | 2.5 | 2.5 |
| Chemical composition | ||
| DM, % of DM | 48.0 | 46.2 |
| CP, % of DM | 17.5 | 19.1 |
| NDF, % of DM | 38.3 | 35.7 |
| ADF, % of DM | 21.5 | 20.5 |
| NFC 3, % of DM | 35.3 | 34.8 |
| Starch, % of DM | 21.7 | 22.4 |
| NEL 4, Mcal/kg of DM | 1.61 | 1.64 |
| Fermentation profile | ||
| pH | 6.01 | 4.69 |
| Lactic acid, % of DM | 0.63 | 8.48 |
| Acetic acid, % of DM | 0.64 | 2.23 |
| NH3-N, % of Total N) | 2.65 | 5.28 |
| Item | Treatment 1 | SEM 2 | p-Value | |
|---|---|---|---|---|
| TMR | FTMR | |||
| pH | 6.50 | 6.41 | 0.10 | 0.55 |
| NH3-N, mg/dL | 13.6 | 21.2 | 0.80 | <0.0001 |
| Total VFA, mM | 78.0 | 90.6 | 2.21 | 0.003 |
| Molar proportion, mmol/100 mmol | ||||
| Acetate | 50.0 | 62.3 | 1.97 | 0.001 |
| Propionate | 18.0 | 19.5 | 0.79 | 0.19 |
| Butyrate | 10.04 | 8.74 | 0.35 | 0.03 |
| Acetate:propionate | 2.80 | 3.22 | 0.17 | 0.12 |
| Item | Phyla and Genera Level | Treatment 1 | SEM 2 | p-Value | |
|---|---|---|---|---|---|
| TMR | FTMR | ||||
| Bacteria | Unclassified_Bacteroidales | 6.00 | 10.47 | 1.16 | 0.02 |
| Verrucomicrobia | 0.60 | 4.87 | 1.17 | 0.03 | |
| Tenericutes | 0.62 | 1.35 | 0.18 | 0.01 | |
| Anaerobic fungi | Ascomycota | 86.8 | 57.0 | 8.97 | 0.04 |
| Candida | 69.9 | 12.9 | 7.87 | 0.0005 | |
| Basidiomycota | 1.52 | 18.1 | 5.03 | 0.04 | |
| Bullera | 0.22 | 2.33 | 0.36 | 0.002 | |
| Cryptococcus | 0.25 | 2.17 | 0.40 | 0.007 | |
| Protozoan | Ostracodinium | 8.37 | 1.45 | 1.55 | 0.01 |
| Archaea | Methanobrevibacter | 92.0 | 95.4 | 1.08 | 0.05 |
| Genus | PH | NH3-N (mg/dL) | Total VFA (mmol/L) | Acetate (%) | Propionate (%) | Butyrate (%) |
|---|---|---|---|---|---|---|
| Unclassified_Bacteroidales | −0.02 | 0.32 | 0.65 * | 0.63 * | 0.43 | −0.41 |
| Candida | 0.17 | −0.42 | −0.71 * | −0.66 * | −0.66 * | 0.27 |
| Bullera | −0.06 | 0.55 | 0.71 * | 0.66 * | 0.59 * | −0.17 |
| Cryptococcus | −0.3 | 0.88 * | 0.56 | 0.64 * | 0.25 | −0.42 |
| Ostracodinium | 0.19 | −0.62 * | −0.76 * | −0.77 * | −0.42 | 0.46 |
| Methanobrevibacter | −0.27 | 0.51 | 0.22 | 0.36 | −0.10 | −0.75 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Ma, Y.; Zhang, H.; Wang, L.; Zhang, Y.; Zhang, G. Fermented Total Mixed Ration Alters Rumen Fermentation Parameters and Microbiota in Dairy Cows. Animals 2023, 13, 1062. https://doi.org/10.3390/ani13061062
Song J, Ma Y, Zhang H, Wang L, Zhang Y, Zhang G. Fermented Total Mixed Ration Alters Rumen Fermentation Parameters and Microbiota in Dairy Cows. Animals. 2023; 13(6):1062. https://doi.org/10.3390/ani13061062
Chicago/Turabian StyleSong, Jiamei, Yuansheng Ma, Hengwei Zhang, Lijun Wang, Yonggen Zhang, and Guangning Zhang. 2023. "Fermented Total Mixed Ration Alters Rumen Fermentation Parameters and Microbiota in Dairy Cows" Animals 13, no. 6: 1062. https://doi.org/10.3390/ani13061062
APA StyleSong, J., Ma, Y., Zhang, H., Wang, L., Zhang, Y., & Zhang, G. (2023). Fermented Total Mixed Ration Alters Rumen Fermentation Parameters and Microbiota in Dairy Cows. Animals, 13(6), 1062. https://doi.org/10.3390/ani13061062