
The Genetic Diversity of Mink (Neovison vison) Populations in China

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Library Construction and Variant Calling
2.3. Genetics Analyses
2.4. Identification of Selective Sweeps
3. Results
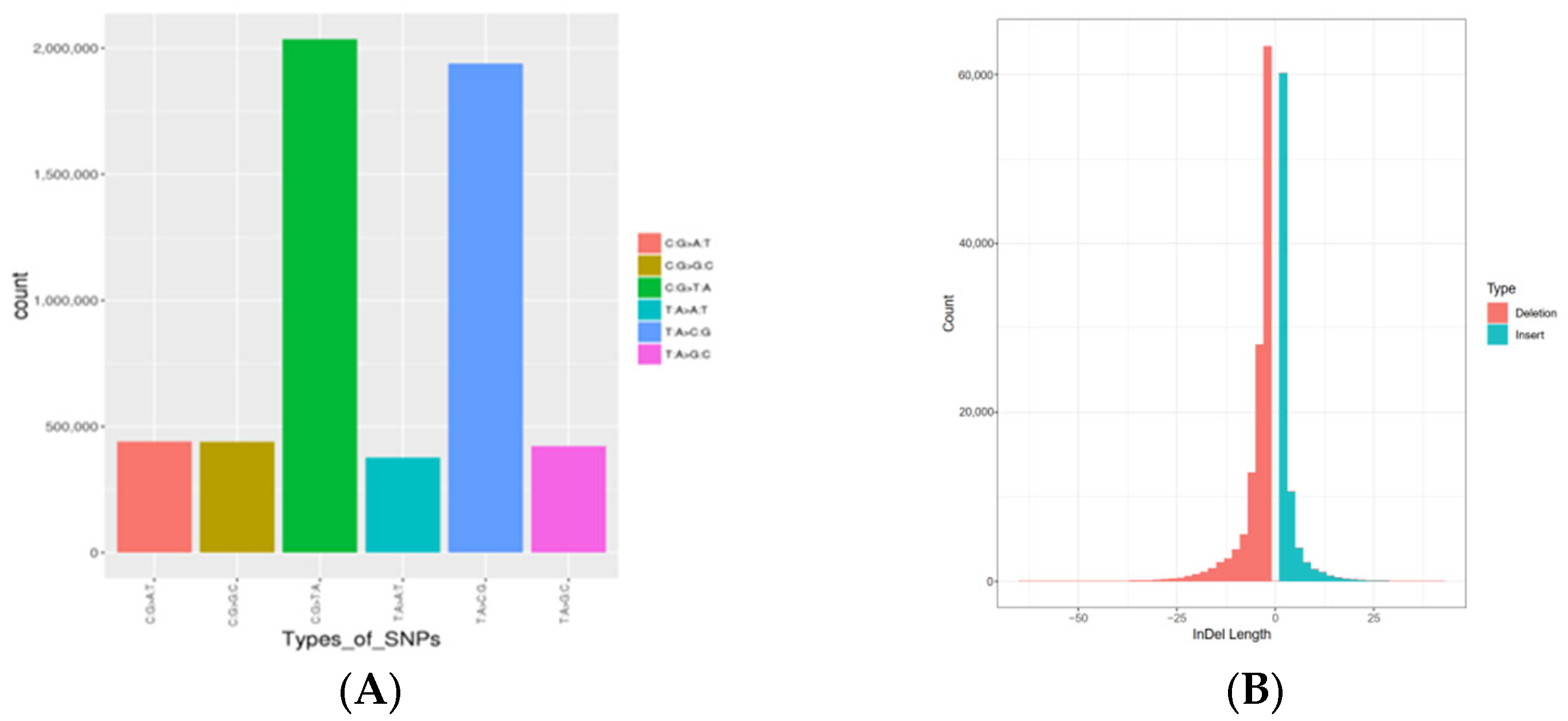
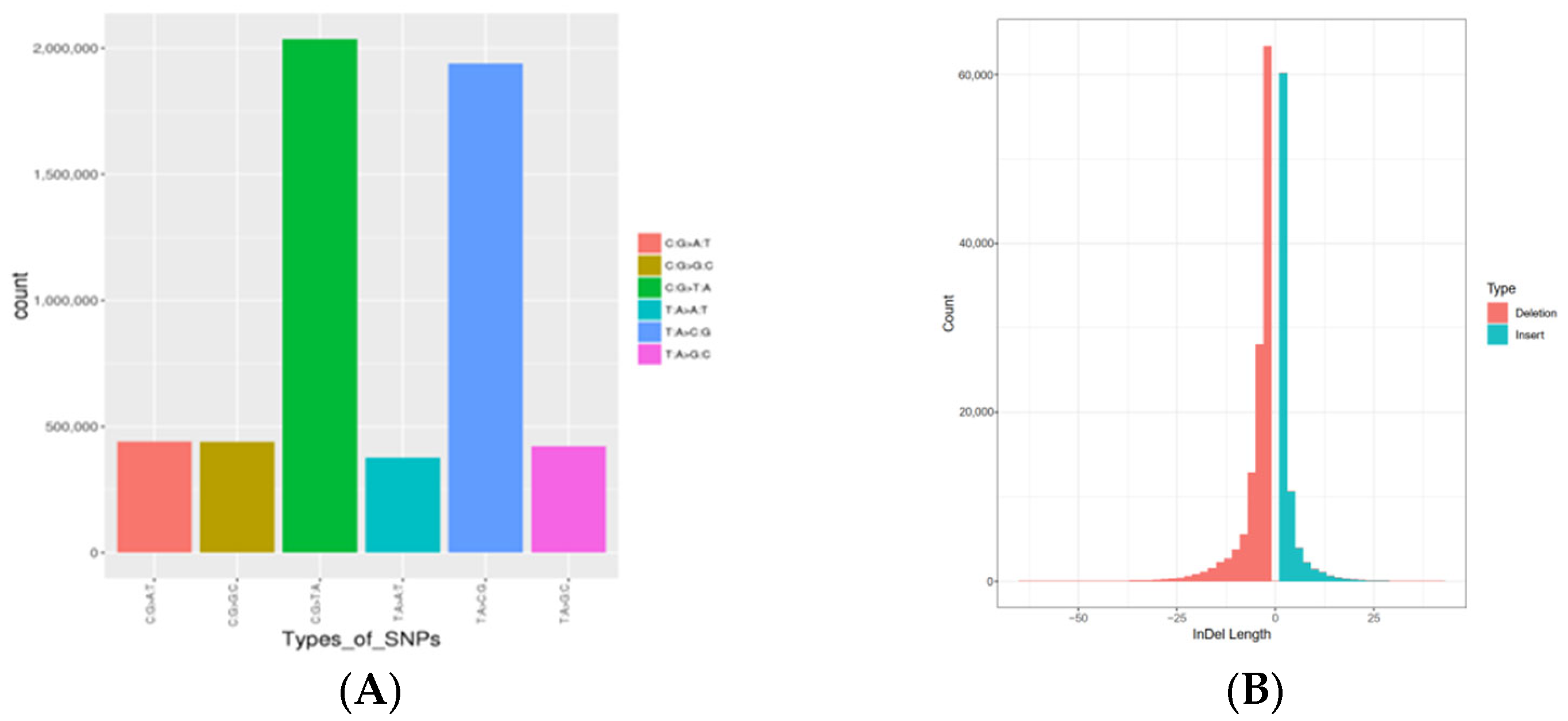
3.1. ddRAD-Seq and Data Filtering
3.2. Variation Calling
3.3. Genetic Diversity
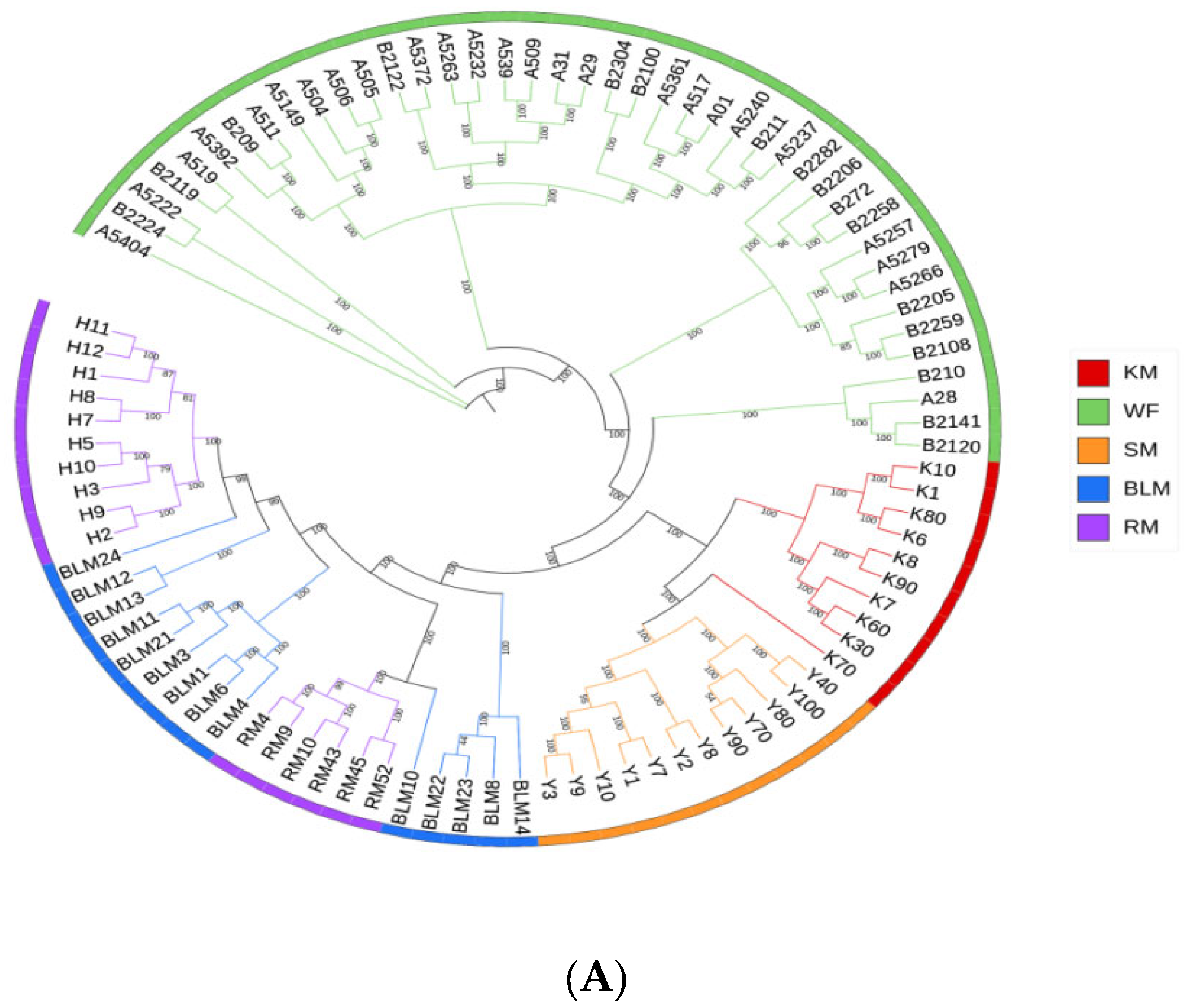
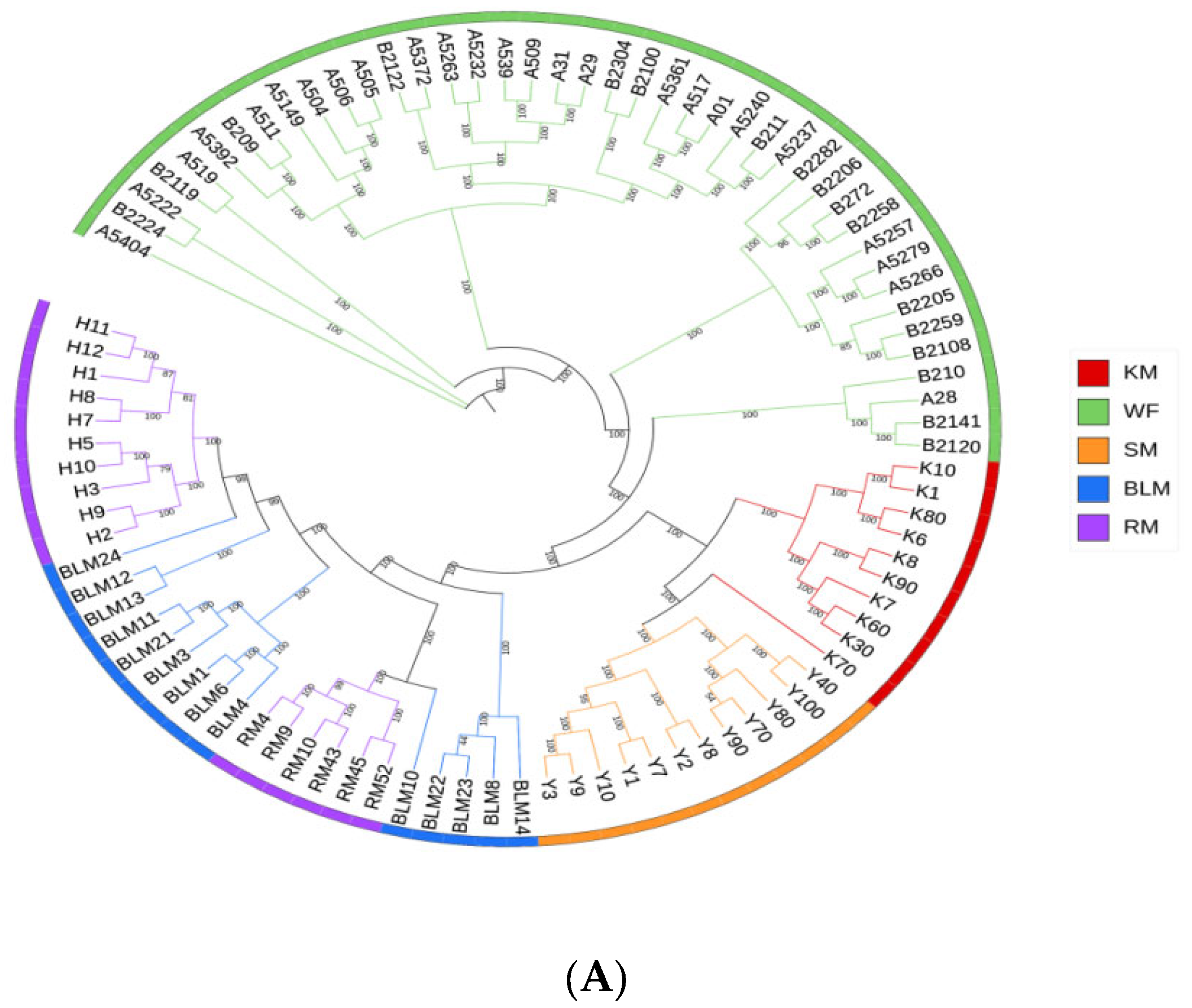
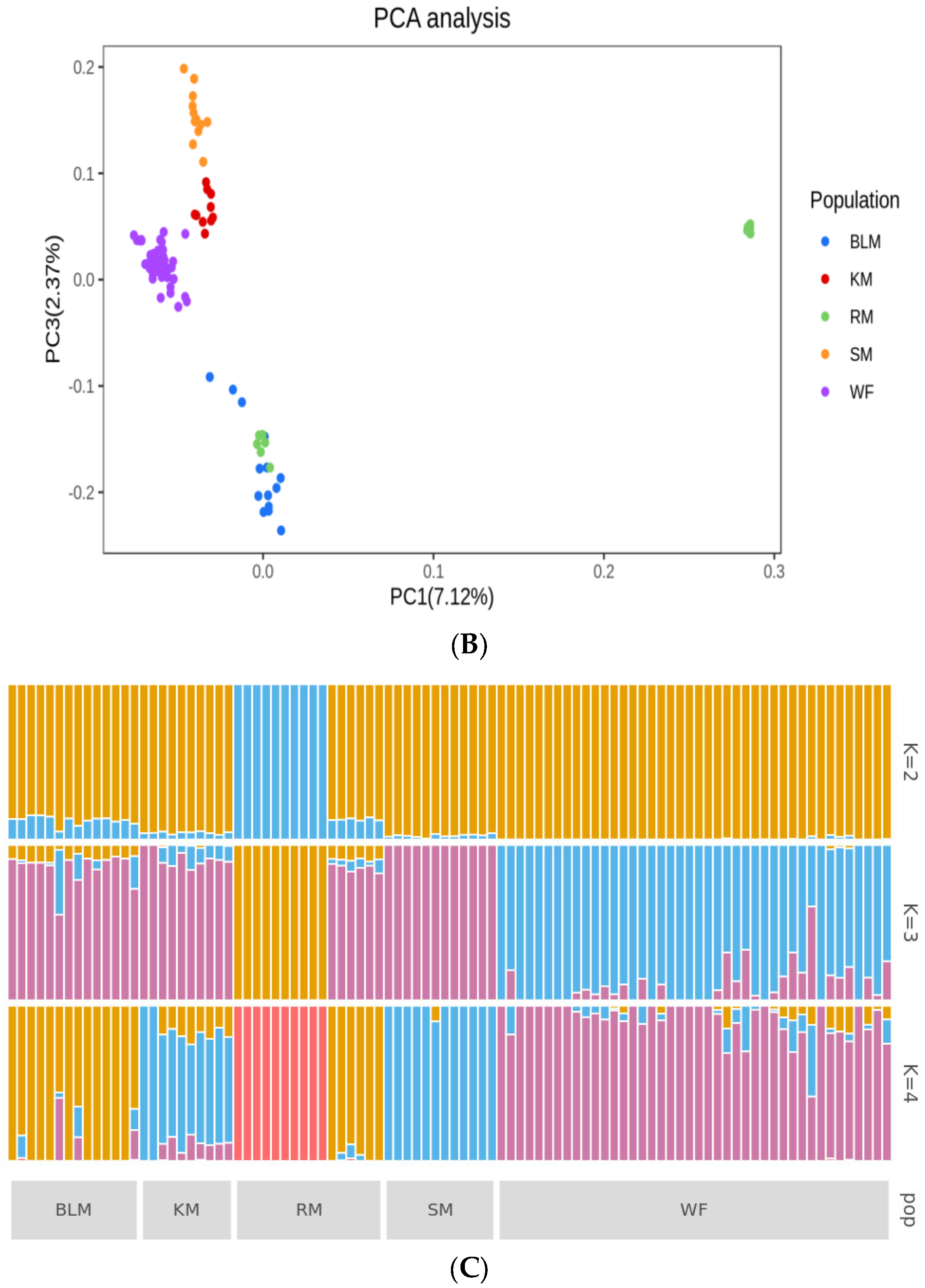
3.4. Genetic Structure
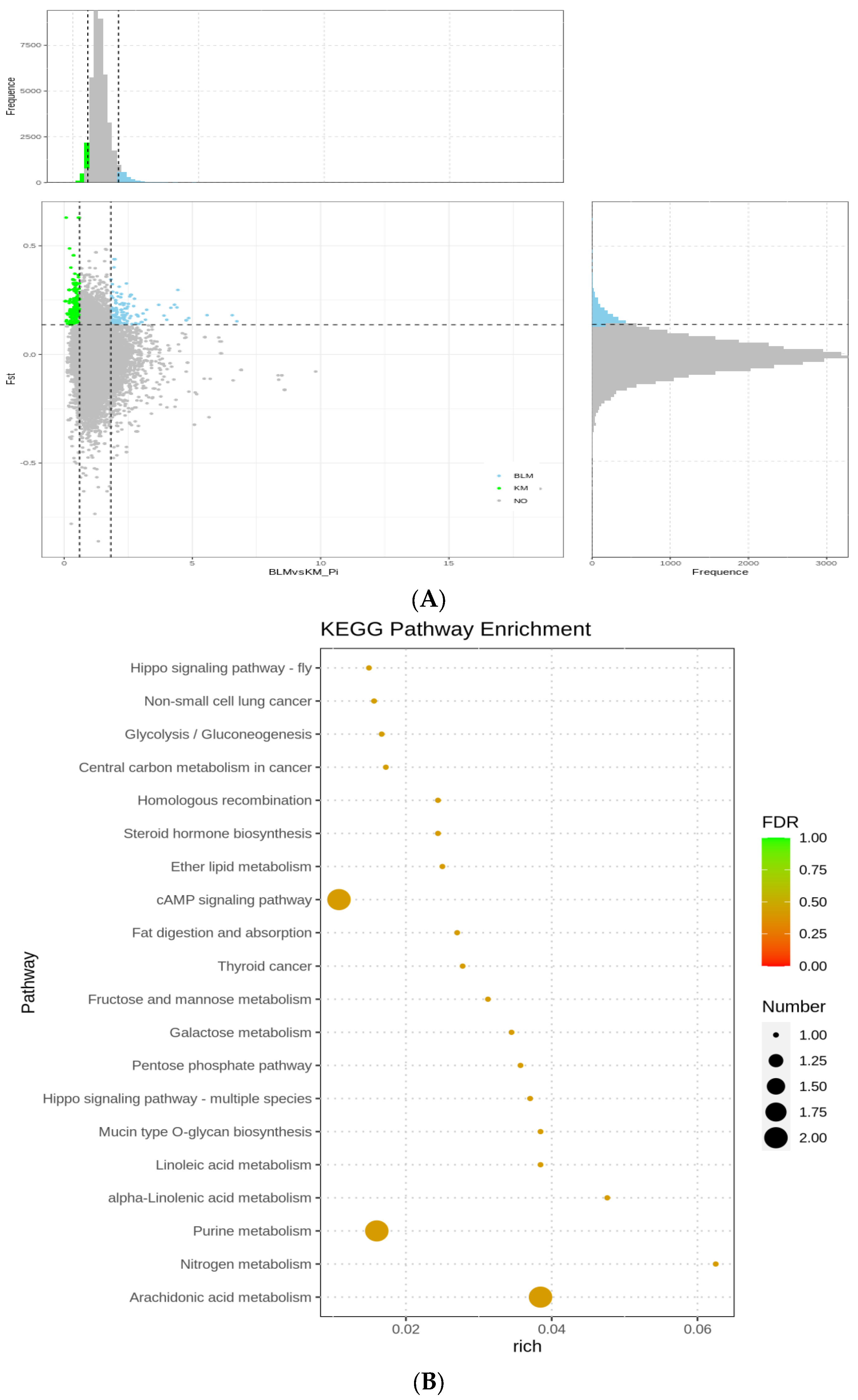
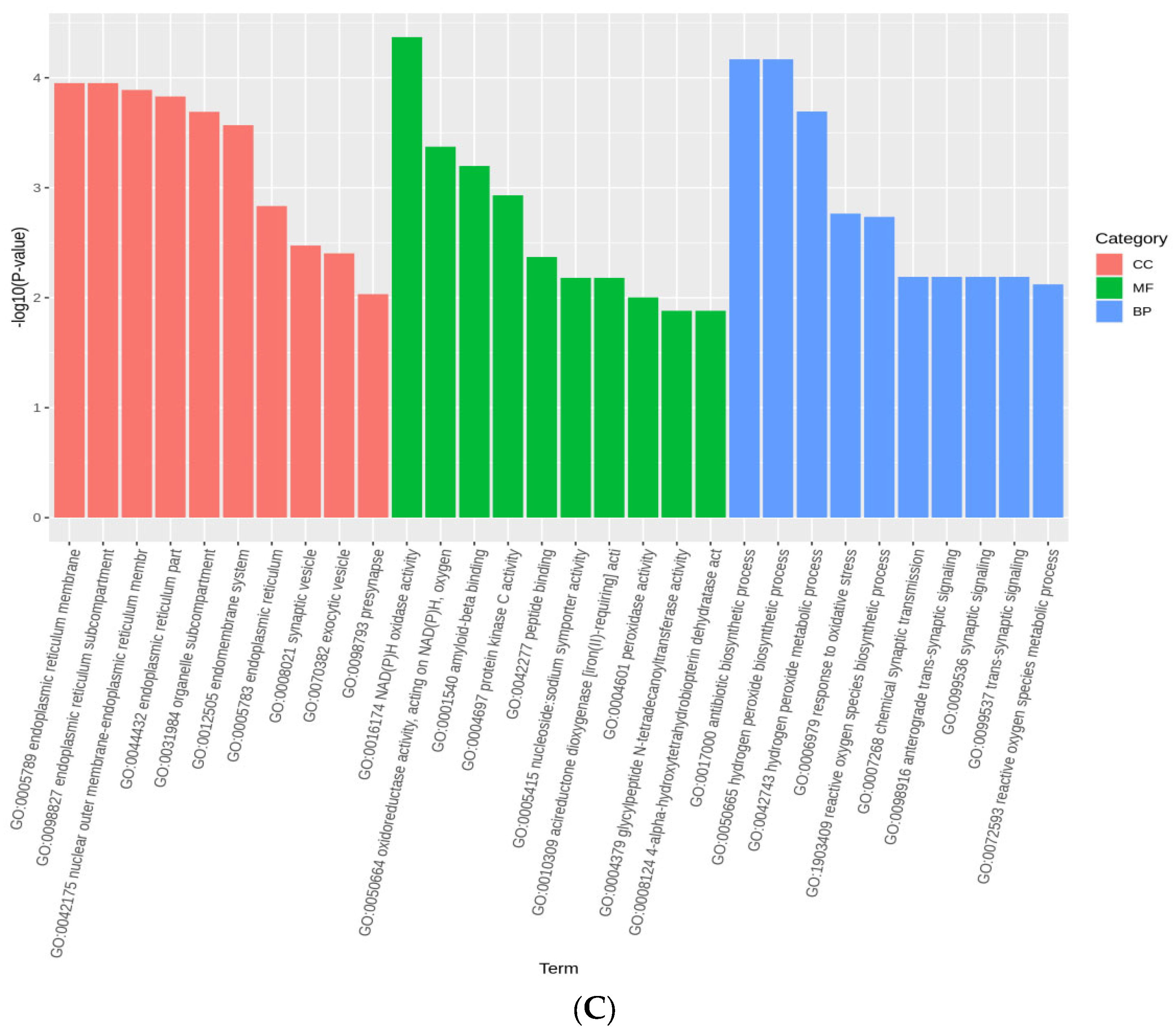
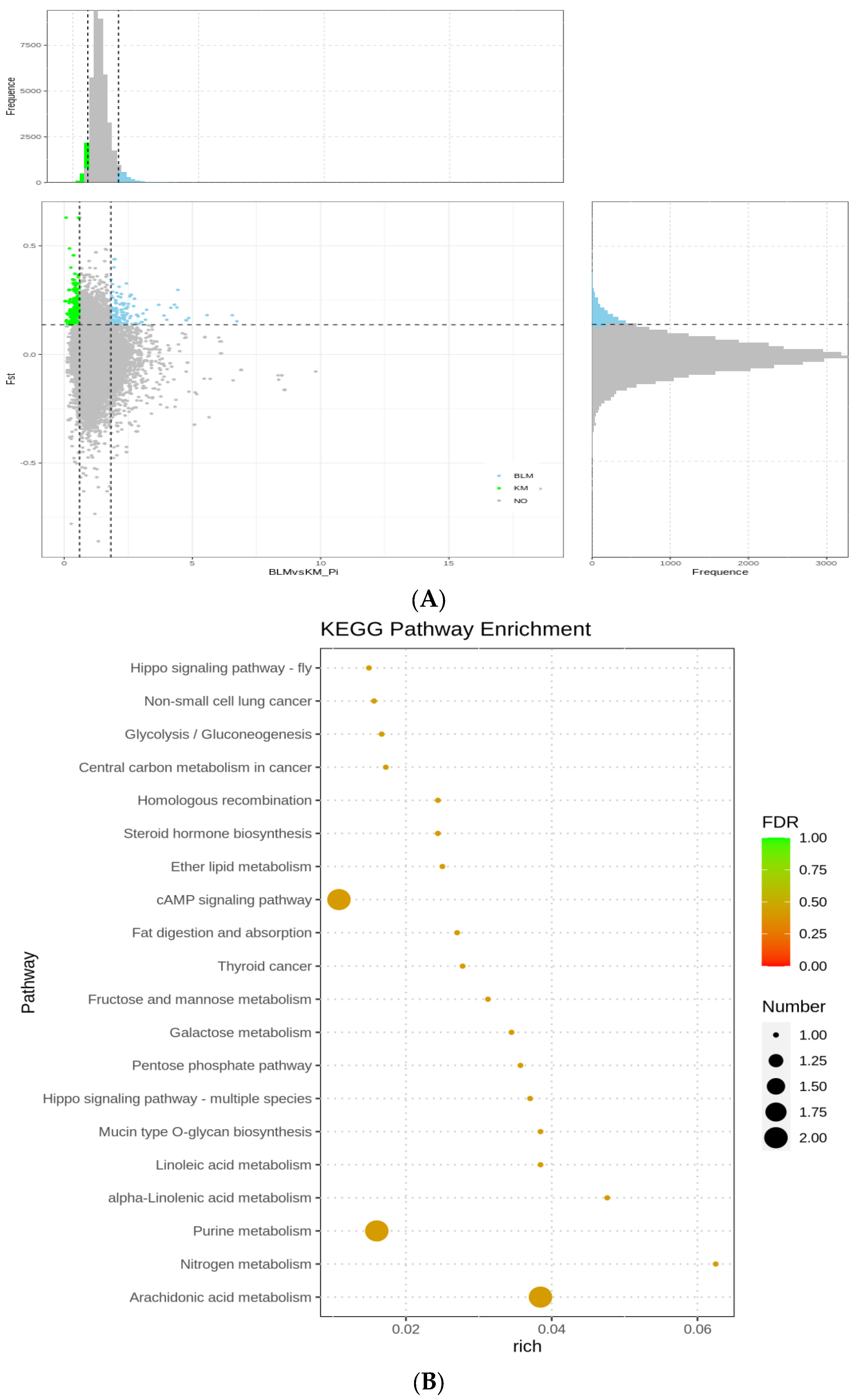
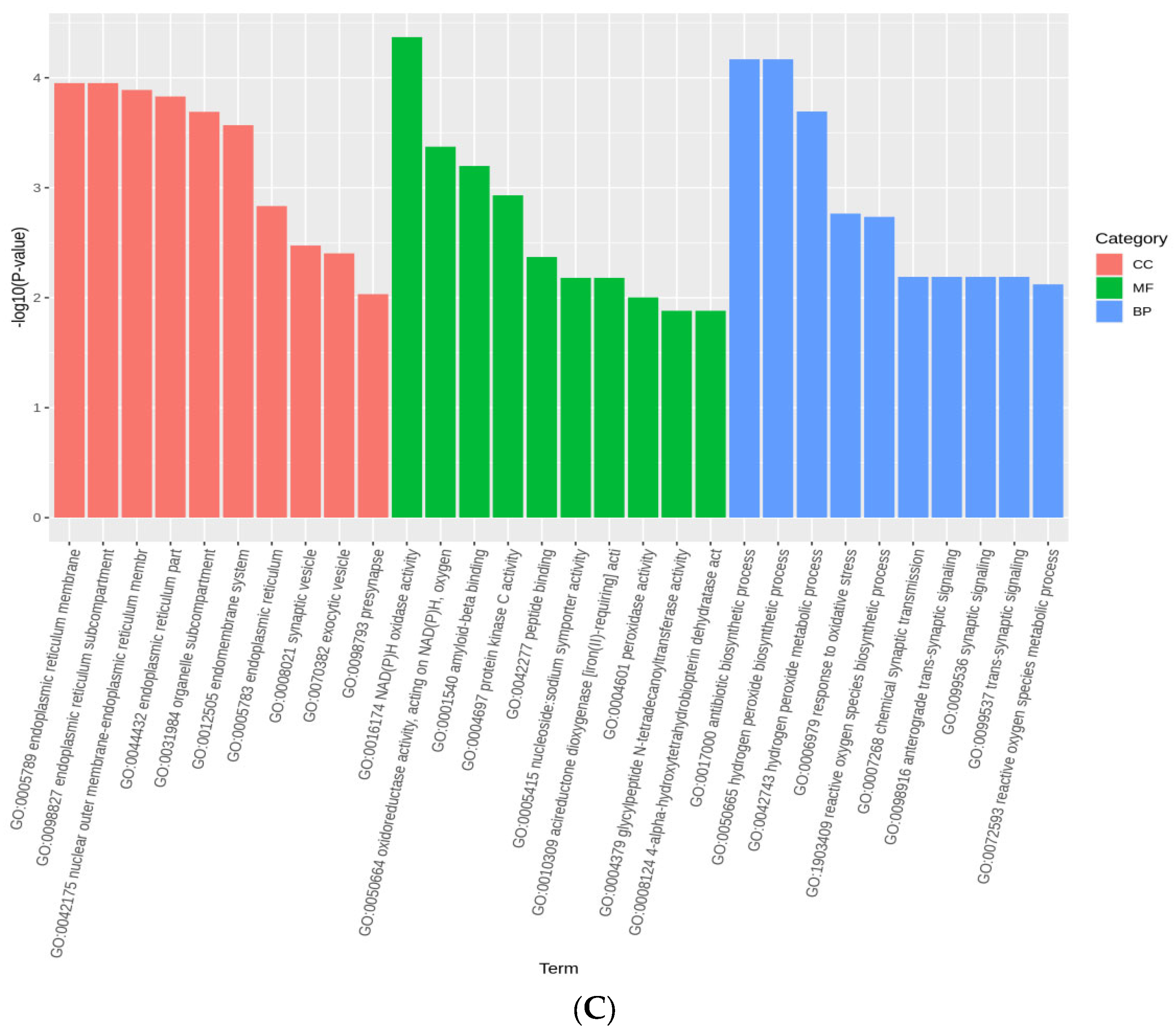
3.5. Selection Signatures and Enrichment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hansen, H.O. The Danish Fur Industry: Structure, Competitiveness and International Position; Djøf: Copenhagen, Denmark, 2016. [Google Scholar]
- Hansen, H.O. The Global Fur Industry: Trends, Globalization and Specialization. J. Agric. Sci. Technol. A 2014, 4, 543–551. [Google Scholar]
- Hansen, H.O. Global Fur Retail Value. Ph.D. Thesis, Department of Food and Resource Economics, University of Copenhagen, Copenhagen, Denmark, 2021. [Google Scholar]
- Ministry of Agriculture and Rural Affairs Published the National Livestock and Poultry Genetic Resources Catalog. Zhejiang Anim. Husb. Vet. Sci. 2020, 45, 32–33.
- Tu, J.; Wang, L.; Xing, X.; Li, Y.; Yang, F. Cloning and Sequence Analysis of Mitochondrial Cytochrome b Gene(Cytb) in Mink(Mustela vison)and Its Evolutionary Relationships with the Mustela. J. Agric. Biotechnol. 2010, 18, 975–980. [Google Scholar]
- Wang, L.; Yang, F. Studies on Correlation of Microsatellite Polymorphism and Economic Character in Mink. Ph.D. Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 2007. Available online: https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C447WN1SO36whK2SkkyaDF9DMFsDdq-V7RsW6e3hmr2gPyhGSzyWS-Gdizz7ygGNGta0na5Ip-7sMbg2CXKoQvAN&uniplatform=NZKPT (accessed on 10 March 2023).
- Yang, H. Study on the Polymorphism of PIT-1 Gene and Genetic Diversity of MinkStudy on the Polymorphism of PIT-1 Gene and Genetic Diversity of Mink; Northeast Agricultural University: Harbin, China, 2010; Available online: https://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CMFD&dbname=CMFD2011&filename=2010264410.nh&v= (accessed on 10 March 2023).
- Shimatani, Y.; Fukue, Y.; Kishimoto, R.; Masuda, R. Genetic Variation and Population Structure of the Feral American Mink (Neovison vison) in Nagano, Japan, Revealed by Microsatellite Analysis. Mammal Study 2010, 35, 1–7. [Google Scholar] [CrossRef]
- Zhang, L.; Hua, Y.; Wei, S. High Genetic Diversity of an Invasive Alien Species: Comparison between Fur-Farmed and Feral American Mink (Neovison vison) in China. Animals 2021, 11, 472. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.; Medina-Vogel, G.; Sepúlveda, M.A.; Noll, D.; Álvarez-Varas, R.; Vianna, J.A. Genetic Structure of Introduced American Mink (Neovison vison) in Patagonia: Colonisation Insights and Implications for Control and Management Strategies. Wildl. Res. 2018, 45, 344–356. [Google Scholar] [CrossRef]
- Zhai, Z.X.; Zhao, W.J.; He, C.; Yang, K.X.; Tang, L.L.; Liu, S.Y.; Zhang, Y.; Huang, Q.Z.; Meng, H. SNPs Discovery and Genotyping Using Restriction-Site-Associated DNA Sequencing(RAD-Seq) in Chickens. Anim. Genet. 2015, 46, 216–219. [Google Scholar] [CrossRef]
- Karimi, K.; Strucken, E.M.; Moghaddar, N.; Ferdosi, M.H.; Esmailizadeh, A.; Gondro, C. Local and Global Patterns of Admixture and Population Structure in Iranian Native Cattle. BMC Genet. 2016, 17, 108. [Google Scholar] [CrossRef]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-Wide Analysis of the World’s Sheep Breeds Reveals High Levels of Historic Mixture and Strong Recent Selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef]
- Colli, L.; Milanesi, M.; Talenti, A.; Bertolini, F.; Chen, M.; Crisà, A.; Daly, K.G.; Del Corvo, M.; Guldbrandtsen, B.; Lenstra, J.A.; et al. Genome-Wide SNP Profiling of Worldwide Goat Populations Reveals Strong Partitioning of Diversity and Highlights Post-Domestication Migration Routes. Genet. Sel. Evol. 2018, 50, 58. [Google Scholar] [CrossRef]
- Edea, Z.; Kim, S.-W.; Lee, K.-T.; Kim, T.H.; Kim, K.-S. Genetic Structure of and Evidence for Admixture between Western and Korean Native Pig Breeds Revealed by Single Nucleotide Polymorphisms. Asian-Australas. J. Anim. Sci. 2014, 27, 1263–1269. [Google Scholar] [CrossRef]
- Cai, Z.; Petersen, B.; Sahana, G.; Madsen, L.B.; Larsen, K.; Thomsen, B.; Bendixen, C.; Lund, M.S.; Guldbrandtsen, B.; Panitz, F. The First Draft Reference Genome of the American Mink (Neovison vison). Sci. Rep. 2017, 7, 14564. [Google Scholar] [CrossRef] [PubMed]
- Miar, Y.; Plastow, G.; Wang, Z.; Sargolzaei, M. PSVII-39 Late-Breaking Abstract: Enhancing Production and Aleutian Disease Resilience in Mink through Advanced Genomics. J. Anim. Sci. 2020, 98, 342. [Google Scholar] [CrossRef]
- Thirstrup, J.P.; Ruiz-Gonzalez, A.; Pujolar, J.M.; Larsen, P.F.; Jensen, J.; Randi, E.; Zalewski, A.; Pertoldi, C. Population Genetic Structure in Farm and Feral American Mink (Neovison vison) Inferred from RAD Sequencing-Generated Single Nucleotide Polymorphisms1. J. Anim. Sci. 2015, 93, 3773–3782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, F.; Cao, J.; Liu, X. Skin Transcriptome Profiles Associated with Skin Color in Chickens. PLoS ONE 2015, 10, e0127301. [Google Scholar] [CrossRef]
- Karimi, K.; Ngoc Do, D.; Sargolzaei, M.; Miar, Y. Population Genomics of American Mink Using Whole Genome Sequencing Data. Genes 2021, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A. Rapid SNP discovery and genetic mapping using sequenced RADmarkers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 March 2023).
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Kai, W.; Li, M.; Hakon, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E.; Orlando, L. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Karimi, K.; Farid, A.H.; Sargolzaei, M.; Myles, S.; Miar, Y. Linkage Disequilibrium, Effective Population Size and Genomic Inbreeding Rates in American Mink Using Genotyping-by-Sequencing Data. Front. Genet. 2020, 11, 223. [Google Scholar] [CrossRef] [PubMed]
- Lohi, O. (Ed.) More about Beautiful Fur Animals—Genetics of Colours, Fur, Defects and Diseases, 1st ed.; NJF Working Group for Fur Anima: Støvring, Denmark.
- Heptner, V.G.; Sludskii, A.A. Mammals of the Soviet Union; Part 1b, Carnivores (Mustelidae and Procyonidae); Smithsonian Institution Libraries and National Science Foundation: Washington, DC, USA, 2002; Volume 2. [Google Scholar]
- Thirstrup, J.P.; Anistoroaei, R.; Guldbrandtsen, B.; Christensen, K.; Fredholm, M.; Nielsen, V.H. Identifying QTL and Genetic Correlations between Fur Quality Traits in Mink (Neovison vison). Anim. Genet. 2014, 45, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.Y.; Bowman, J.; Schulte-Hostedde, A.; Wilson, P.J. Functional Genetic Diversity of Domestic and Wild American Mink (Neovison vison). Evol. Appl. 2020, 13, 2610–2629. [Google Scholar] [CrossRef] [PubMed]
- Price, E. Behavioral Aspects of Animal Domestication. Q. Rev. Biol. 1984, 59, 1–32. [Google Scholar] [CrossRef]
- Cieslak, M.; Reissmann, M.; Hofreiter, M.; Ludwig, A. Colours of Domestication. Biol. Rev. Camb. Philos. Soc. 2011, 86, 885–899. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Liu, L.L.; Song, X.C.; Cong, B.; Yang, F.H. Heritability and Genetic Trends for Growth and Fur Quality Traits in Silver Blue Mink. Ital. J. Anim. Sci. 2016, 16, 39–43. [Google Scholar] [CrossRef]
- Li, Y.X.; Zhang, Q.; Shang, X.M.; Zhang, Y.; Liu, C.Q.; Song, J.D.; Yue, B.C. Effect of Shenshao oral lotion to serum lipid and oxidative injury in atherosclerotic rats. J. North China Univ. Sci. Technol. Health Sci. Ed. 2017, 19, 457–461. [Google Scholar] [CrossRef]
- Wu, X.; Hammer, J.A. Melanosome Transfer: It Is Best to Give and Receive. Curr. Opin. Cell Biol. 2014, 29, 1–7. [Google Scholar] [CrossRef]
- Sheng, W.; Weng, S.; Li, F.; Zhang, Y.; Liu, K. Immunohistological Localization of Mel1a Melatonin Receptor in Pigeon Retina. Nat. Sci. Sleep 2021, 13, 113–121. [Google Scholar] [CrossRef]
- Baxter, L.L.; Watkins-Chow, D.E.; Pavan, W.J.; Loftus, S.K. A Curated Gene List for Expanding the Horizons of Pigmentation Biology. Pigment Cell Melanoma Res. 2019, 32, 348–358. [Google Scholar] [CrossRef]
- Mikkola ML: Genetic basis of skin appendage development. Semin. Cell. Dev. Biol. 2007, 18, 225–236. [CrossRef] [PubMed]
- Ho, H.; Kapadia, R.; Al-Tahan, S.; Ahmad, S.; Ganesan, A.K. WIPI1 Coordinates Melanogenic Gene Transcription and Melanosome Formation via TORC1 Inhibition. J. Biol. Chem 2011, 286, 12509–12523. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viennet, C.; Robin, S.; Berthon, J.-Y.; He, L.; Humbert, P. Precise Role of Dermal Fibroblasts on Melanocyte Pigmentation. J. Dermatol. Sci. 2017, 88, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Rishikaysh, P.; Dev, K.; Diaz, D.; Qureshi, W.M.S.; Filip, S.; Mokry, J. Signaling Involved in Hair Follicle Morphogenesis and Development. Int. J. Mol. Sci. 2014, 15, 1647–1670. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhang, Y.; Xing, Y.; Xu, W.; Guo, H.; Deng, F.; Ma, X.; Li, Y. The Balance of Bmp6 and Wnt10b Regulates the Telogen-Anagen Transition of Hair Follicles. Cell Commun. Signal. 2019, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Markakis, M.N.; Soedring, V.E.; Dantzer, V.; Christensen, K.; Anistoroaei, R. Association of MITF Gene with Hearing and Pigmentation Phenotype in Hedlund White American Mink (Neovison vison). J. Genet. 2014, 93, 1–5. [Google Scholar] [CrossRef]
- Anistoroaei, R.; Fredholm, M.; Christensen, K.; Leeb, T. Albinism in the American Mink (Neovison vison) Is Associated with a Tyrosinase Nonsense Mutation. Anim. Genet. 2010, 39, 645–648. [Google Scholar] [CrossRef]
- Cirera, S.; Markakis, M.N.; Christensen, K.; Anistoroaei, R. New Insights into the Melanophilin (MLPH) Gene Controlling Coat Color Phenotypes in American Mink. Gene 2013, 527, 48–54. [Google Scholar] [CrossRef]
- Anistoroaei, R.; Krogh, A.K.; Christensen, K. A Frameshift Mutation in the LYST Gene Is Responsible for the Aleutian Color and the Associated Chédiak–Higashi Syndrome in American Mink. Anim. Genet. 2013, 44, 178–183. [Google Scholar] [CrossRef]
- Cirera, S.; Markakis, M.N.; Kristiansen, T.; Vissenberg, K.; Fredholm, M.; Christensen, K.; Anistoroaei, R. A Large Insertion in Intron 2 of the TYRP1 Gene Associated with American Palomino Phenotype in American Mink. Mamm. Genome 2016, 27, 135–143. [Google Scholar] [CrossRef]
- Manakhov, A.D.; Mintseva, M.Y.; Andreev, I.A.; Uralsky, L.I.; Andreeva, T.V.; Trapezov, O.V.; Rogaev, E.I. Genome Analysis of American Minks Reveals Link of Mutations in Ras-Related Protein-38 Gene to Moyle Brown Coat Phenotype. Sci. Rep. 2020, 10, 15876. [Google Scholar] [CrossRef] [PubMed]
- Manakhov, A.D.; Mintseva, M.Y.; Andreeva, T.V.; Trapezov, O.V.; Rogaev, E.I. Shadow Coat Colour in American Mink Associated with a Missense Mutation in the KIT Gene. Anim. Genet. 2022, 53, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Manakhov, A.D.; Mintseva, M.Y.; Uralsky, L.I.; Andreeva, T.V.; Trapezov, O.V.; Rogaev, E.I. Identification of Mutant Gene for Black Crystal Coat and Non-Allelic Gene Interactions in Neogale Vison. Sci. Rep. 2022, 12, 10483. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Average Sequencing Depth | Coverage | Coverage at Least 4X | Coverage at Least 10X | Coverage at Least 20X |
|---|---|---|---|---|---|
| WM | 1.44 | 7.44% | 4.79% | 3.32% | 2.21% |
| BLM | 1.66 | 8.56% | 5.59% | 3.91% | 2.56% |
| BM | 1.56 | 7.88% | 5.06% | 3.56% | 2.38% |
| RM | 1.54 | 8.08% | 5.24% | 3.69% | 2.43% |
| SM | 1.49 | 7.11% | 4.48% | 3.13% | 2.22% |
| Population | HOM_REF | HET | UNKNOWN | HOM_ALT | SUM |
|---|---|---|---|---|---|
| WM | 200,812 | 49,808 | 594,182 | 76,330 | 38,687,586 |
| BLM | 225,361 | 67,709 | 542,191 | 85,870 | 12,895,862 |
| BM | 218,001 | 50,891 | 574,606 | 77,633 | 92,11,330 |
| RM | 234,273 | 65,438 | 533,296 | 88,124 | 14,738,128 |
| SM | 203,639 | 40,290 | 602,817 | 74,386 | 11,053,596 |
| Total | 1,082,086 | 274,136 | 2,847,092 | 402,343 | 86,586,502 |
| Type | All | |
|---|---|---|
| Number | Percentage | |
| exonic | 36,286 | 2.6 |
| synonymous SNV | 19,444 | 1.39 |
| nonsynonymous SNV | 16,420 | 1.18 |
| stop-gain | 382 | 0.03 |
| stop-loss | 27 | 0 |
| unknown | 13 | 0 |
| splicing | 261 | 0.02 |
| ncRNA total | 0 | 0 |
| ncRNA exonic | 0 | 0 |
| ncRNA splicing | 0 | 0 |
| ncRNA exonic; splicing | 0 | 0 |
| ncRNA intronic | 0 | 0 |
| intronic | 491,190 | 35.18 |
| intergenic | 829,525 | 59.41 |
| UTR5 | 1990 | 0.14 |
| UTR3 | 5321 | 0.38 |
| UTR5; UTR3 | 4 | 0 |
| upstream | 15,222 | 1.09 |
| downstream | 16,057 | 1.15 |
| upstream; downstream | 401 | 0.03 |
| Total | 1,396,257 | 100 |
| Population | Obs Het | Obs Hom | Exp Het | Exp Hom | π | Fis |
|---|---|---|---|---|---|---|
| BLM | 0.1317 | 0.7683 | 0.2725 | 0.7275 | 0.3503 | 0.2641 |
| BM | 0.105 | 0.895 | 0.1971 | 0.8029 | 0.2209 | 0.2517 |
| RM | 0.1167 | 0.8833 | 0.1928 | 0.8072 | 0.2099 | 0.2441 |
| SM | 0.1006 | 0.8994 | 0.1961 | 0.8039 | 0.2261 | 0.2786 |
| WM | 0.1089 | 0.8911 | 0.2552 | 0.7448 | 0.2731 | 0.4402 |
| BM | RM | SM | WM | |
|---|---|---|---|---|
| BLM | 0.134487 | 0.148431 | 0.148311 | 0.110624 |
| BM | 0.154566 | 0.136017 | 0.083326 | |
| RM | 0.16579 | 0.11867 | ||
| SM | 0.096049 |
| Coat Color Name | Symbol | Gene | Reference |
|---|---|---|---|
| STANDRAD | |||
| White types | [43,44] | ||
| Albino | cc | MITF | |
| Hedlund | hh | TYR | |
| Grey types | |||
| Sliverblue | pp | MLPH | |
| Steelblue | psps | [45,46] | |
| Aleutian | aa | LYST | |
| Brown types | |||
| Palomino | kk | TYRP1 | [47,48] |
| Moyle buff | mm | RAB38 | |
| Patterned | |||
| Goofus | oo | ||
| Others | |||
| Shadow | SRSR | KIT | [49] |
| Black crystal | Cr Cr | COPA | [50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Li, H.; Larsen, P.F.; Ba, H.; Shi, H.; Zhang, H.; Liu, Z. The Genetic Diversity of Mink (Neovison vison) Populations in China. Animals 2023, 13, 1497. https://doi.org/10.3390/ani13091497
Zhang T, Li H, Larsen PF, Ba H, Shi H, Zhang H, Liu Z. The Genetic Diversity of Mink (Neovison vison) Populations in China. Animals. 2023; 13(9):1497. https://doi.org/10.3390/ani13091497
Chicago/Turabian StyleZhang, Tietao, Hu Li, Peter Foged Larsen, Hengxing Ba, Hongyu Shi, Haihua Zhang, and Zongyue Liu. 2023. "The Genetic Diversity of Mink (Neovison vison) Populations in China" Animals 13, no. 9: 1497. https://doi.org/10.3390/ani13091497
APA StyleZhang, T., Li, H., Larsen, P. F., Ba, H., Shi, H., Zhang, H., & Liu, Z. (2023). The Genetic Diversity of Mink (Neovison vison) Populations in China. Animals, 13(9), 1497. https://doi.org/10.3390/ani13091497