
A Study of the Resistance of Hu Sheep Lambs to Escherichia coli F17 Based on Whole Genome Sequencing

 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Sample Collection
2.3. Sheep Blood Genomic DNA Extraction and Sequencing
2.4. Sequencing Data Filtering
2.5. Reference Genome Matching
2.6. SNP Mutation Detection
2.7. Genome-Wide Association Analysis
2.8. GO and KEGG Enrichment Analysis
2.9. Protein Interaction Network Analysis
3. Results
3.1. Quality Control of whole Genome Sequencing Data of Antagonistic and Susceptible Hu Sheep Lambs
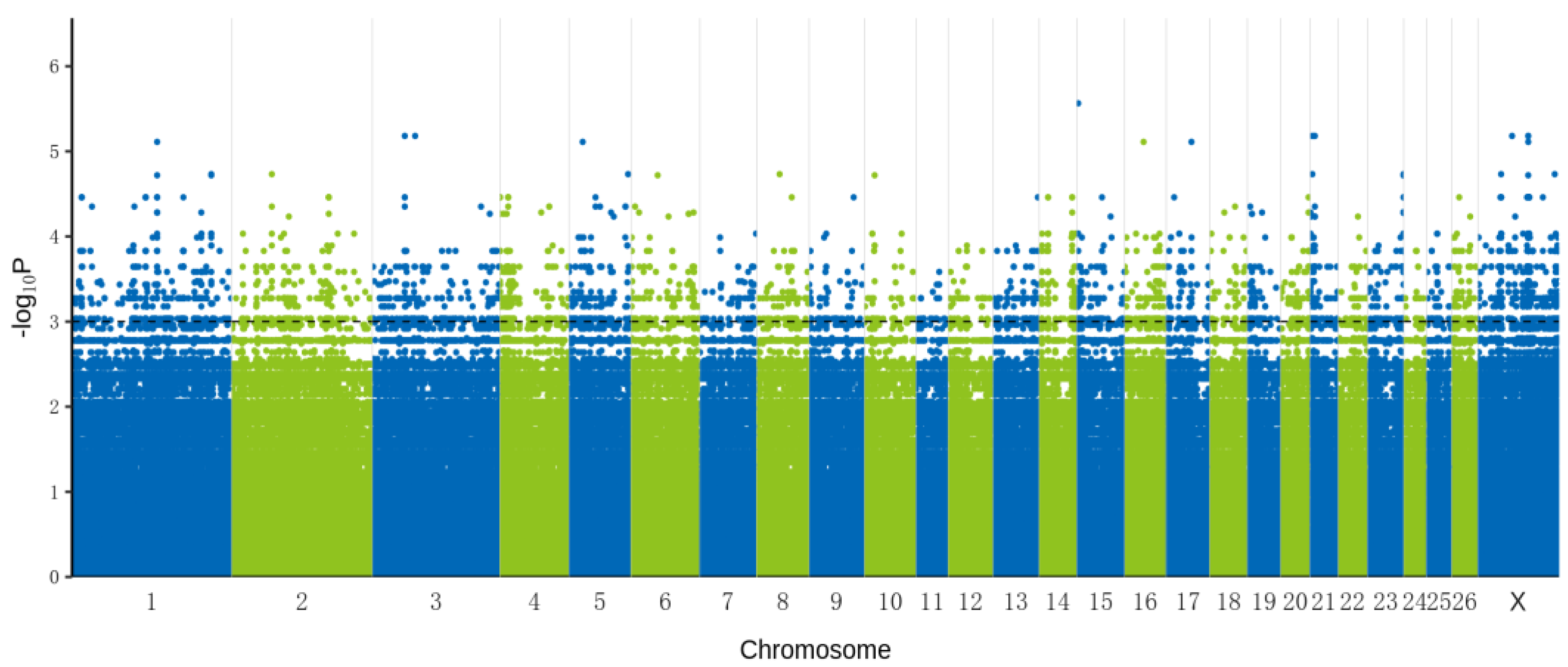
3.2. Genome-Wide Association Analysis
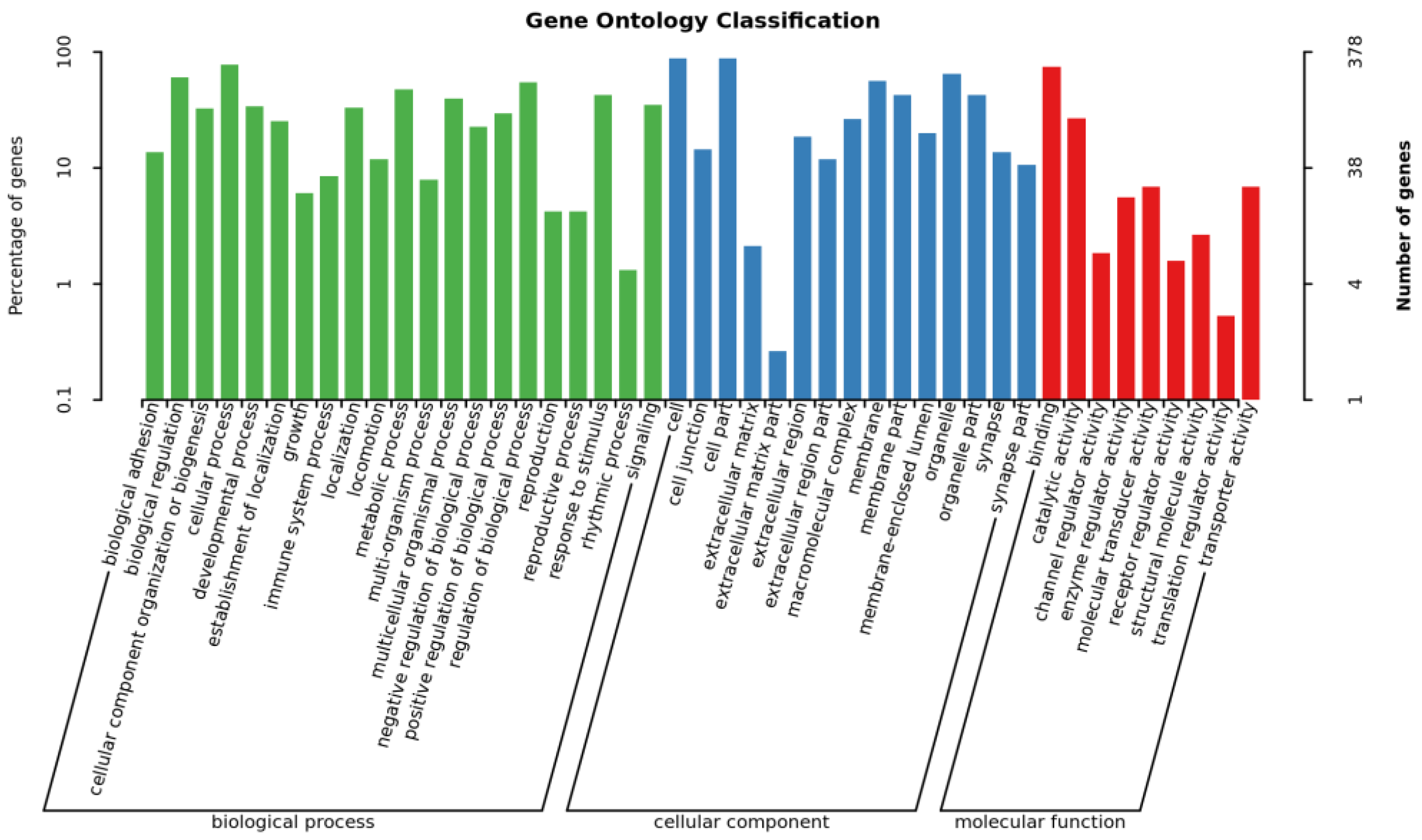
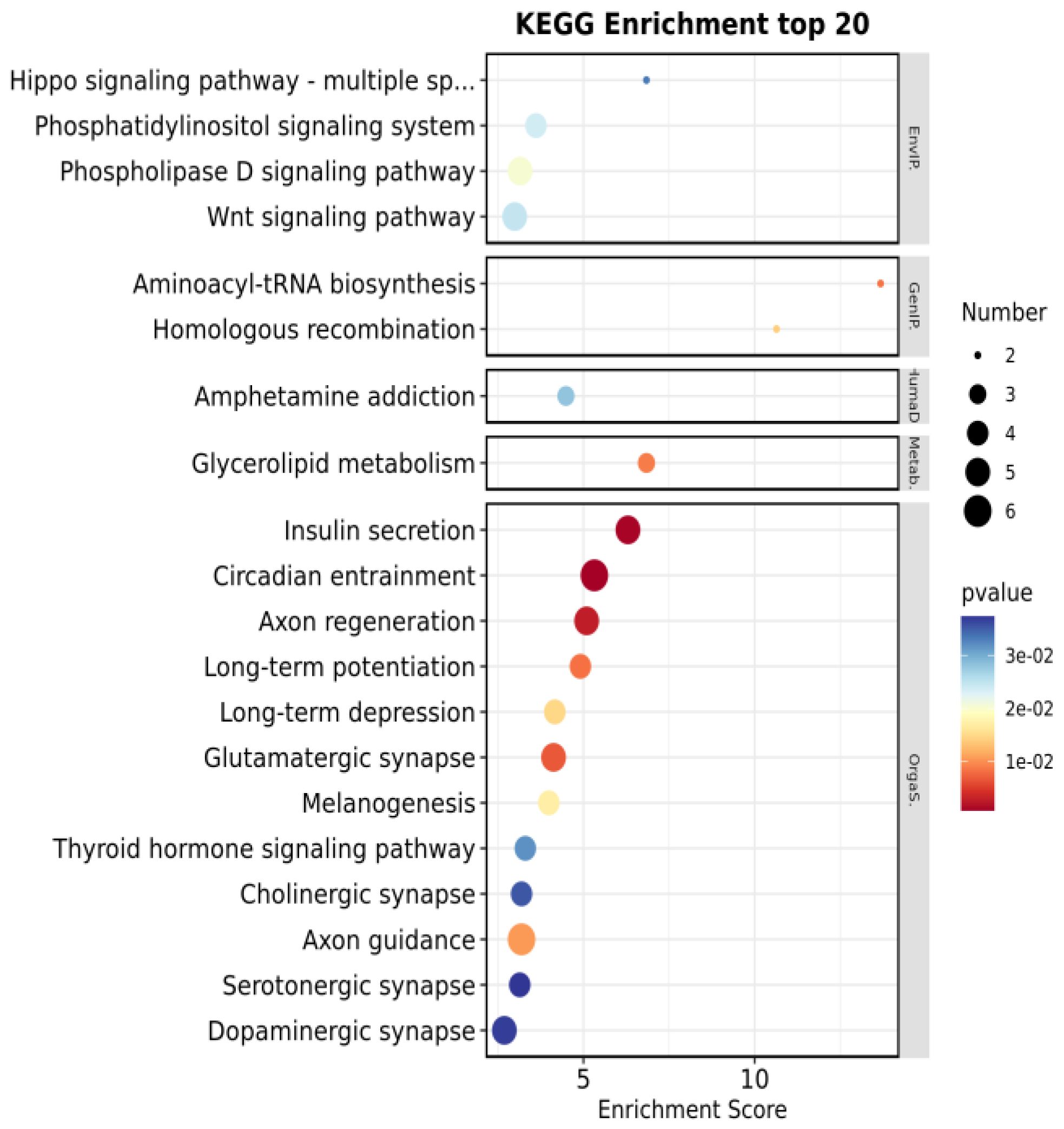
3.3. GO and KEGG Enrichment Analysis
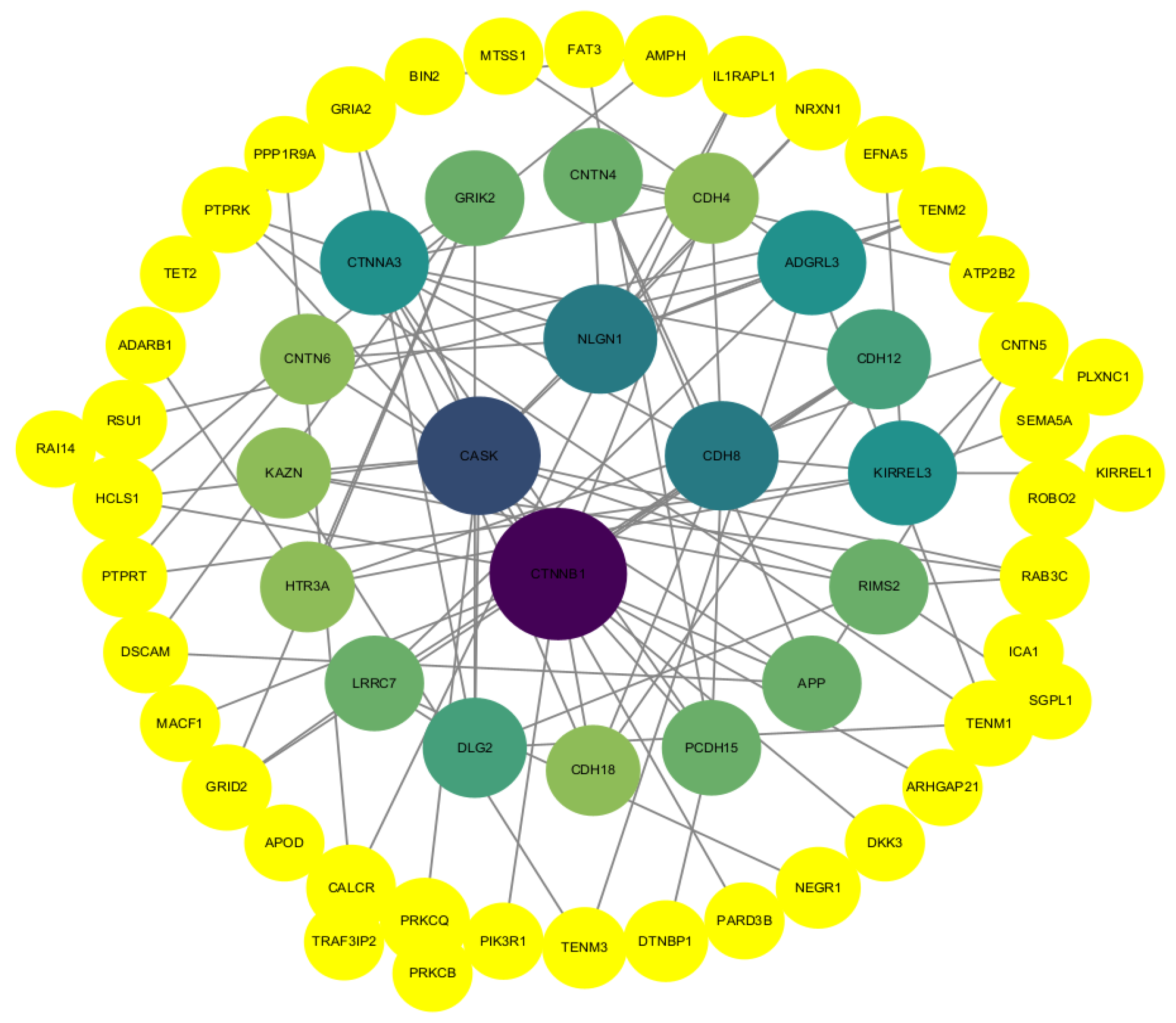
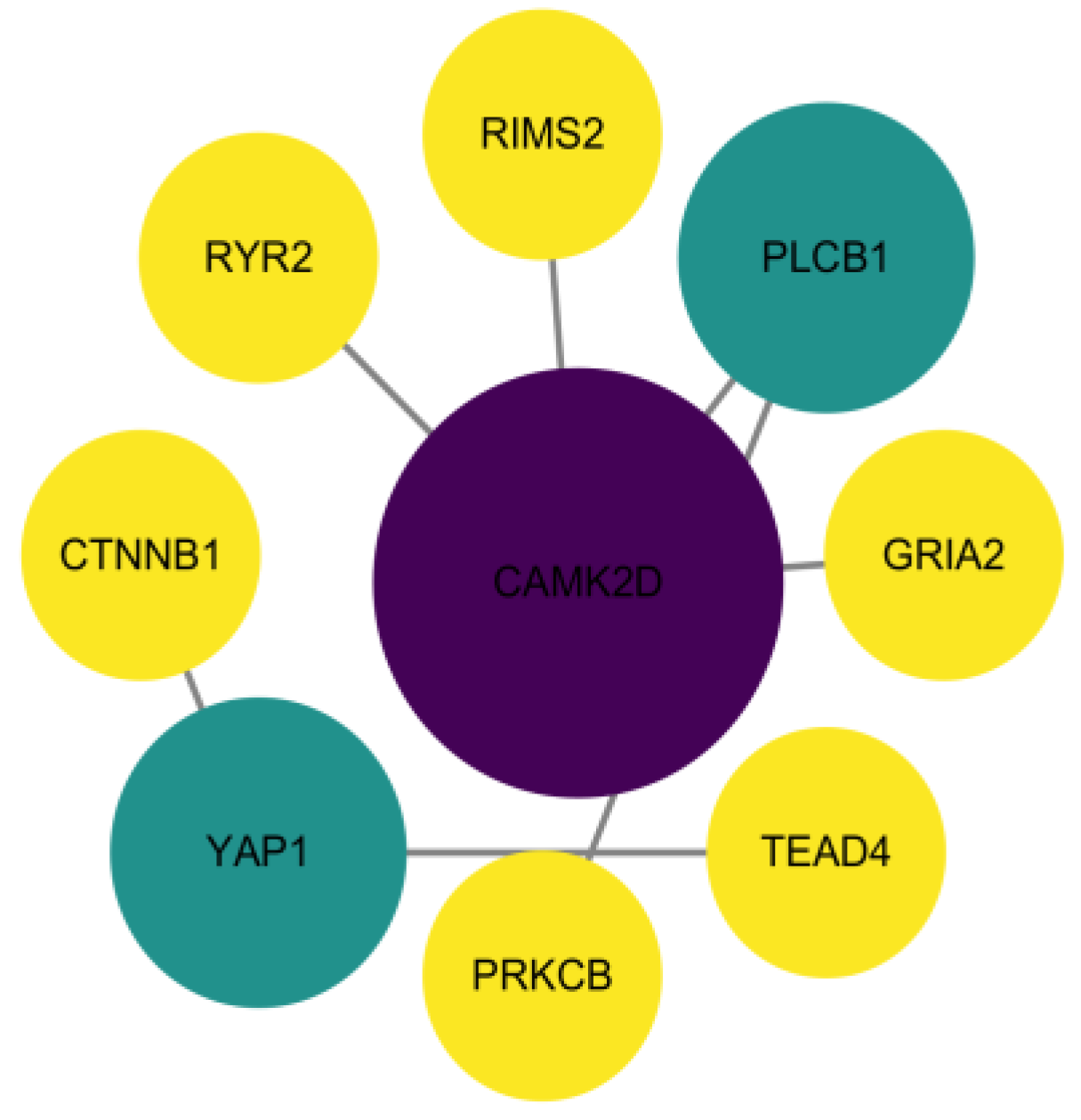
3.4. Protein Interaction Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, X.; Zhao, H.; Zhou, Z.; Miao, Y.; Li, R.; Yang, B.; Cao, C.; Xiao, S.; Wang, X.; Liu, H.; et al. Characterisation of Extended-Spectrum β-Lactamase-Producing Escherichia coli Isolates That Cause Diarrhea in Sheep in Northwest China. Microbiol. Spectr. 2022, 10, e0159522. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Le Bouguénec, C.; Bertin, Y. AFA and F17 adhesins produced by pathogenic Escherichia coli strains in domestic animals. Vet. Res. 1999, 30, 317–342. [Google Scholar] [PubMed]
- Lintermans, P.F.; Pohl, P.; Bertels, A.; Charlier, G.; Vandekerckhove, J.; Van Damme, J.; Schoup, J.; Schlicker, C.; Korhonen, T.; De Greve, H.; et al. Characterization and purification of the F17 adhesin on the surface of bovine enteropathogenic and septicemic Escherichia coli. Am. J. Vet. Res. 1988, 49, 1794–1799. [Google Scholar]
- El Mazouari, K.; Oswald, E.; Hernalsteens, J.P.; Lintermans, P.; De Greve, H. F17-like fimbriae from an invasive Escherichia coli strain producing cytotoxic necrotizing factor type 2 toxin. Infect. Immun. 1994, 62, 2633–2638. [Google Scholar] [CrossRef]
- Bertin, Y.; Girardeau, J.P.; Darfeuille-Michaud, A.; Contrepois, M. Characterization of 20K fimbria, a new adhesin of septicemic and diarrhea-associated Escherichia coli strains, that belongs to a family of adhesins with N-acetyl-D-glucosamine recognition. Infect. Immun. 1996, 64, 332–342. [Google Scholar] [CrossRef]
- Bertin, Y.; Martin, C.; Oswald, E.; Girardeau, J.P. Rapid and specific detection of F17-related pilin and adhesin genes in diarrheic and septicemic Escherichia coli strains by multiplex PCR. J. Clin. Microbiol. 1996, 34, 2921–2928. [Google Scholar] [CrossRef]
- Bihannic, M.; Ghanbarpour, R.; Auvray, F.; Cavalié, L.; Châtre, P.; Boury, M.; Brugère, H.; Madec, J.Y.; Oswald, E. Identification and detection of three new F17 fimbrial variants in Escherichia coli strains isolated from cattle. Vet. Res. 2014, 45, 76. [Google Scholar] [CrossRef]
- Cid, D.; Sanz, R.; Marín, I.; de Greve, H.; Ruiz-Santa-Quiteria, J.A.; Amils, R.; de la Fuente, R. Characterization of nonenterotoxigenic Escherichia coli strains producing F17 fimbriae isolated from diarrheic lambs and goat kids. J. Clin. Microbiol. 1999, 37, 1370–1375. [Google Scholar] [CrossRef]
- Osman, K.; Mustafa, A.; Elhariri, M.; Abdelhamed, G.J.T.; diseases, e. The distribution of Escherichia coli serovars, virulence genes, gene association and combinations and virulence genes encoding serotypes in pathogenic E. coli recovered from diarrhoeic calves, sheep and goat. Transbound. Emerg. Dis. 2013, 60, 69–78. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, P.; Zhao, Y.; Ma, X. Enterotoxigenic Escherichia coli: Intestinal pathogenesis mechanisms and colonization resistance by gut microbiota. Gut Microbes 2022, 14, 2055943. [Google Scholar] [CrossRef] [PubMed]
- Kagnoff, M.F.; Eckmann, L. Epithelial cells as sensors for microbial infection. J. Clin. Investig. 1997, 100, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Sun, Y.; Cui, H.; Zhu, S.J.; Qiu, H.J. Mucosal vaccines: Strategies and challenges. Immunol. Lett. 2020, 217, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Gu, K.; Wang, F.; Jia, G.; Zhao, H.; Chen, X.; Wu, C.; Zhang, R.; Tian, G.; Cai, J.; et al. Tryptophan Ameliorates Barrier Integrity and Alleviates the Inflammatory Response to Enterotoxigenic Escherichia coli K88 Through the CaSR/Rac1/PLC-γ1 Signaling Pathway in Porcine Intestinal Epithelial Cells. Front. Immunol. 2021, 12, 748497. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, P.; Wang, J.; Xu, J.; Liu, C.; Qiao, H.; Gan, L.; Duan, E.; Zhang, Y.; Wang, M.; et al. Zinc Laurate Protects against Intestinal Barrier Dysfunction and Inflammation Induced by ETEC in a Mice Model. Nutrients 2022, 15, 54. [Google Scholar] [CrossRef]
- Peng, H.; Shen, Y.; Zhang, Q.; Liu, J.; Wang, Z.; Huang, L.; Zhou, F.; Yu, J.; Liu, M.; Yuan, Y.; et al. Qihuang decoction promotes the recovery of intestinal immune barrier dysfunction after gastrectomy in rats. Am. J. Transl. Res. 2018, 10, 827–836. [Google Scholar]
- Hao, W.; Hao, C.; Wu, C.; Xu, Y.; Jin, C. Aluminum induced intestinal dysfunction via mechanical, immune, chemical and biological barriers. Chemosphere 2022, 288, 132556. [Google Scholar] [CrossRef]
- Chen, W.; Lv, X.; Zhang, W.; Hu, T.; Cao, X.; Ren, Z.; Getachew, T.; Mwacharo, J.M.; Haile, A.; Sun, W. Insights Into Long Non-Coding RNA and mRNA Expression in the Jejunum of Lambs Challenged With Escherichia coli F17. Front. Vet. Sci. 2022, 9, 819917. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Sahana, G.; Guldbrandtsen, B.; Bendixen, C.; Lund, M.S. Genome-wide association mapping for female fertility traits in Danish and Swedish Holstein cattle. Anim. Genet. 2010, 41, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.J.B. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv 2014. bioRxiv:005165. [Google Scholar]
- Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Silva-García, O.; Valdez-Alarcón, J.J.; Baizabal-Aguirre, V.M. Wnt/β-Catenin Signaling as a Molecular Target by Pathogenic Bacteria. Front. Immunol. 2019, 10, 2135. [Google Scholar] [CrossRef]
- Chan, H.; Li, Q.; Wang, X.; Liu, W.Y.; Hu, W.; Zeng, J.; Xie, C.; Kwong, T.N.Y.; Ho, I.H.T.; Liu, X.; et al. Vitamin D(3) and carbamazepine protect against Clostridioides difficile infection in mice by restoring macrophage lysosome acidification. Autophagy 2022, 18, 2050–2067. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhou, J.; Chen, X.; Zhou, Y.; Song, X.; Cai, B.; Zhang, J.; Lu, X.; Ying, B. Pathway Analyses Identify Novel Variants in the WNT Signaling Pathway Associated with Tuberculosis in Chinese Population. Sci. Rep. 2016, 6, 28530. [Google Scholar] [CrossRef] [PubMed]
- Queirós, J.; Alves, P.C.; Vicente, J.; Gortázar, C.; de la Fuente, J. Genome-wide associations identify novel candidate loci associated with genetic susceptibility to tuberculosis in wild boar. Sci. Rep. 2018, 8, 1980. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, A.; Katayama, Y.; Kakegawa, W.; Ino, D.; Nishiyama, M.; Yuzaki, M.; Nakayama, K.I. The autism-associated protein CHD8 is required for cerebellar development and motor function. Cell Rep. 2021, 35, 108932. [Google Scholar] [CrossRef] [PubMed]
- Pagnamenta, A.T.; Khan, H.; Walker, S.; Gerrelli, D.; Wing, K.; Bonaglia, M.C.; Giorda, R.; Berney, T.; Mani, E.; Molteni, M.; et al. Rare familial 16q21 microdeletions under a linkage peak implicate cadherin 8 (CDH8) in susceptibility to autism and learning disability. J. Med. Genet. 2011, 48, 48–54. [Google Scholar] [CrossRef]
- Frei, J.A.; Niescier, R.F.; Bridi, M.S.; Durens, M.; Nestor, J.E.; Kilander, M.B.C.; Yuan, X.; Dykxhoorn, D.M.; Nestor, M.W.; Huang, S.; et al. Regulation of Neural Circuit Development by Cadherin-11 Provides Implications for Autism. eNeuro 2021, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, M.; Kracht, S.S.; Esteso, G.; Cirera, S.; Edfors, I.; Archibald, A.L.; Bendixen, C.; Andersson, L.; Fredholm, M.; Jørgensen, C.B. Refined candidate region specified by haplotype sharing for Escherichia coli F4ab/F4ac susceptibility alleles in pigs. Anim. Genet. 2010, 41, 21–25. [Google Scholar] [CrossRef]
- Rossi, U.A.; Hasenauer, F.C.; Caffaro, M.E.; Neumann, R.; Salatin, A.; Poli, M.A.; Rossetti, C.A. A haplotype at intron 8 of PTPRT gene is associated with resistance to Brucella infection in Argentinian creole goats. Vet. Microbiol. 2017, 207, 133–137. [Google Scholar] [CrossRef]
- Aytekin, C.; Germeshausen, M.; Tuygun, N.; Tanir, G.; Dogu, F.; Ikinciogullari, A. Kostmann disease with developmental delay in three patients. Eur. J. Pediatr. 2010, 169, 759–762. [Google Scholar] [CrossRef]
- Qin, W.; Brands, X.; Van’t Veer, C.; de Vos, A.F.; Scicluna, B.P.; van der Poll, T. Bronchial Epithelial Tet2 Maintains Epithelial Integrity during Acute Pseudomonas aeruginosa Pneumonia. Infect. Immun. 2020, 89, 10-1128. [Google Scholar] [CrossRef]
- Zhu, C.; Cai, Y.; Mo, S.; Zhu, J.; Wang, W.; Peng, B.; Guo, J.; Zhang, Z.; Chen, X. NF-κB-mediated TET2-dependent TNF promoter demethylation drives Mtb-upregulation TNF expression in macrophages. Tuberculosis 2021, 129, 102108. [Google Scholar] [CrossRef] [PubMed]
- Maddugoda, M.P.; Stefani, C.; Gonzalez-Rodriguez, D.; Saarikangas, J.; Torrino, S.; Janel, S.; Munro, P.; Doye, A.; Prodon, F.; Aurrand-Lions, M.; et al. cAMP signaling by anthrax edema toxin induces transendothelial cell tunnels, which are resealed by MIM via Arp2/3-driven actin polymerization. Cell Host Microbe 2011, 10, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.F., 2nd; Behrendt, C.L.; Ruhn, K.A.; Lee, S.; Raj, P.; Takahashi, J.S.; Hooper, L.V. The microbiota coordinates diurnal rhythms in innate immunity with the circadian clock. Cell 2021, 184, 4154–4167.e12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, K.; Hu, Z.; Yang, L.; Wei, B.; Li, S.; Qin, X.; Yang, P.; Yu, H. SIRT5 is important for bacterial infection by regulating insulin secretion and glucose homeostasis. Protein Cell 2020, 11, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xie, S.; Miao, J.; Li, Y.; Wang, Z.; Wang, M.; Yu, Q. Lactobacillus reuteri maintains intestinal epithelial regeneration and repairs damaged intestinal mucosa. Gut Microbes 2020, 11, 997–1014. [Google Scholar] [CrossRef]
- Rogan, M.R.; Patterson, L.L.; Wang, J.Y.; McBride, J.W. Bacterial Manipulation of Wnt Signaling: A Host-Pathogen Tug-of-Wnt. Front. Immunol. 2019, 10, 2390. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sjölinder, M.; Wang, X.; Altenbacher, G.; Hagner, M.; Berglund, P.; Gao, Y.; Lu, T.; Jonsson, A.B.; Sjölinder, H. Thyroid hormone enhances nitric oxide-mediated bacterial clearance and promotes survival after meningococcal infection. PLoS ONE 2012, 7, e41445. [Google Scholar] [CrossRef]
- Chen, Y.; Jing, H.; Chen, M.; Liang, W.; Yang, J.; Deng, G.; Guo, M. Transcriptional Profiling of Exosomes Derived from Staphylococcus aureus-Infected Bovine Mammary Epithelial Cell Line MAC-T by RNA-Seq Analysis. Oxidative Med. Cell. Longev. 2021, 2021, 8460355. [Google Scholar] [CrossRef]
- Ma, Y.C.; Yang, Z.S.; Ma, L.Q.; Shu, R.; Zou, C.G.; Zhang, K.Q. YAP in epithelium senses gut barrier loss to deploy defenses against pathogens. PLoS Pathog. 2020, 16, e1008766. [Google Scholar] [CrossRef]
- LaCanna, R.; Liccardo, D.; Zhang, P.; Tragesser, L.; Wang, Y.; Cao, T.; Chapman, H.A.; Morrisey, E.E.; Shen, H.; Koch, W.J.; et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Investig. 2019, 129, 2107–2122. [Google Scholar] [CrossRef]
- Salhi, I.; Rabti, A.; Dhehibi, A.; Raouafi, N. Sandwich-Based Immunosensor for Dual-Mode Detection of Pathogenic F17-Positive Escherichia coli Strains. Int. J. Mol. Sci. 2022, 23, 6028. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lv, X.; Zhang, W.; Hu, T.; Cao, X.; Ren, Z.; Getachew, T.; Mwacharo, J.M.; Haile, A.; Sun, W. Non-Coding Transcriptome Provides Novel Insights into the Escherichia coli F17 Susceptibility of Sheep Lamb. Biology 2022, 11, 348. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Cao, Y.; Shan, H.; Wu, J.; Song, X.; Jiang, Y. The GWAS Analysis of Body Size and Population Verification of Related SNPs in Hu Sheep. Front. Genet. 2021, 12, 642552. [Google Scholar] [CrossRef] [PubMed]
- Bolormaa, S.; Swan, A.A.; Stothard, P.; Khansefid, M.; Moghaddar, N.; Duijvesteijn, N.; van der Werf, J.H.J.; Daetwyler, H.D.; MacLeod, I.M. A conditional multi-trait sequence GWAS discovers pleiotropic candidate genes and variants for sheep wool, skin wrinkle and breech cover traits. Genet. Sel. Evol. 2021, 53, 58. [Google Scholar] [CrossRef]
- Gebreselassie, G.; Berihulay, H.; Jiang, L.; Ma, Y. Review on Genomic Regions and Candidate Genes Associated with Economically Important Production and Reproduction Traits in Sheep (Ovies aries). Animals 2019, 10, 33. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | R-R | C-R | C-R-P | R-B | C-B | C-B-P | G-C | >Q20 | >Q30 |
|---|---|---|---|---|---|---|---|---|---|
| s2S-250 | 328124474 | 323466836 | 98.58% | 49.22G | 48.38G | 98.30% | 43.32% | 97.56% | 93.05% |
| s2S-936 | 316593058 | 311927296 | 98.53% | 47.49G | 46.66G | 98.26% | 43.47% | 97.52% | 92.96% |
| s2S-088 | 280099462 | 275608400 | 98.40% | 42.01G | 41.22G | 98.10% | 43.24% | 97.08% | 91.80% |
| s2S-037 | 291558274 | 286959356 | 98.42% | 43.73G | 42.89G | 98.06% | 43.41% | 97.30% | 92.28% |
| s2S-341 | 298812648 | 293506672 | 98.22% | 44.82G | 43.84G | 97.80% | 43.63% | 97.13% | 91.87% |
| s2S-940 | 292608542 | 288114728 | 98.46% | 43.89G | 43.05G | 98.08% | 43.58% | 97.24% | 92.08% |
| s2A-926 | 229463262 | 225940978 | 98.46% | 34.42G | 33.80G | 98.21% | 43.48% | 97.55% | 93.06% |
| s2A-965 | 251932032 | 248203718 | 98.52% | 37.79G | 37.10G | 98.17% | 43.40% | 97.69% | 93.38% |
| s2A-349 | 253327108 | 249116262 | 98.34% | 38.00G | 37.22G | 97.94% | 43.76% | 97.19% | 92.02% |
| s2A-187 | 266199676 | 261823920 | 98.36% | 39.93G | 39.13G | 98.01% | 43.37% | 97.14% | 91.90% |
| s2A-982 | 247628212 | 243590354 | 98.37% | 37.14G | 36.40G | 97.99% | 43.60% | 97.23% | 92.22% |
| s2A-937 | 229237666 | 225756938 | 98.48% | 34.39G | 33.69G | 97.98% | 43.09% | 97.45% | 92.68% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, Y.; Su, P.; Gu, Y.; Lv, X.; Cao, X.; Wang, S.; Yuan, Z.; Sun, W. A Study of the Resistance of Hu Sheep Lambs to Escherichia coli F17 Based on Whole Genome Sequencing. Animals 2024, 14, 161. https://doi.org/10.3390/ani14010161
Duan Y, Su P, Gu Y, Lv X, Cao X, Wang S, Yuan Z, Sun W. A Study of the Resistance of Hu Sheep Lambs to Escherichia coli F17 Based on Whole Genome Sequencing. Animals. 2024; 14(1):161. https://doi.org/10.3390/ani14010161
Chicago/Turabian StyleDuan, Yanjun, Pengwei Su, Yifei Gu, Xiaoyang Lv, Xiukai Cao, Shanhe Wang, Zehu Yuan, and Wei Sun. 2024. "A Study of the Resistance of Hu Sheep Lambs to Escherichia coli F17 Based on Whole Genome Sequencing" Animals 14, no. 1: 161. https://doi.org/10.3390/ani14010161
APA StyleDuan, Y., Su, P., Gu, Y., Lv, X., Cao, X., Wang, S., Yuan, Z., & Sun, W. (2024). A Study of the Resistance of Hu Sheep Lambs to Escherichia coli F17 Based on Whole Genome Sequencing. Animals, 14(1), 161. https://doi.org/10.3390/ani14010161