
Descriptive Statistics and Genome-Wide Copy Number Analysis of Milk Production Traits of Jiangsu Chinese Holstein Cows


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Phenotypic Data Collection and Quality Control
2.3. Analysis of Phenotypic and Genotypic Parameters
2.4. Genotyping Data and Quality Control
2.5. Population Structure Analysis
2.6. Detection of CNVR
2.7. Association Analysis Based on CNVR
2.8. Bioinformatics Analysis of Candidate Genes
3. Results
3.1. Descriptive Statistics of Milk Production Traits of the Chinese Holstein Cattle in Jiangsu
3.2. Phenotypic and Genetic Correlation among the Different Parities’ Milk Production Traits of the Dairy Cows in Jiangsu
3.3. Group Structure Analysis
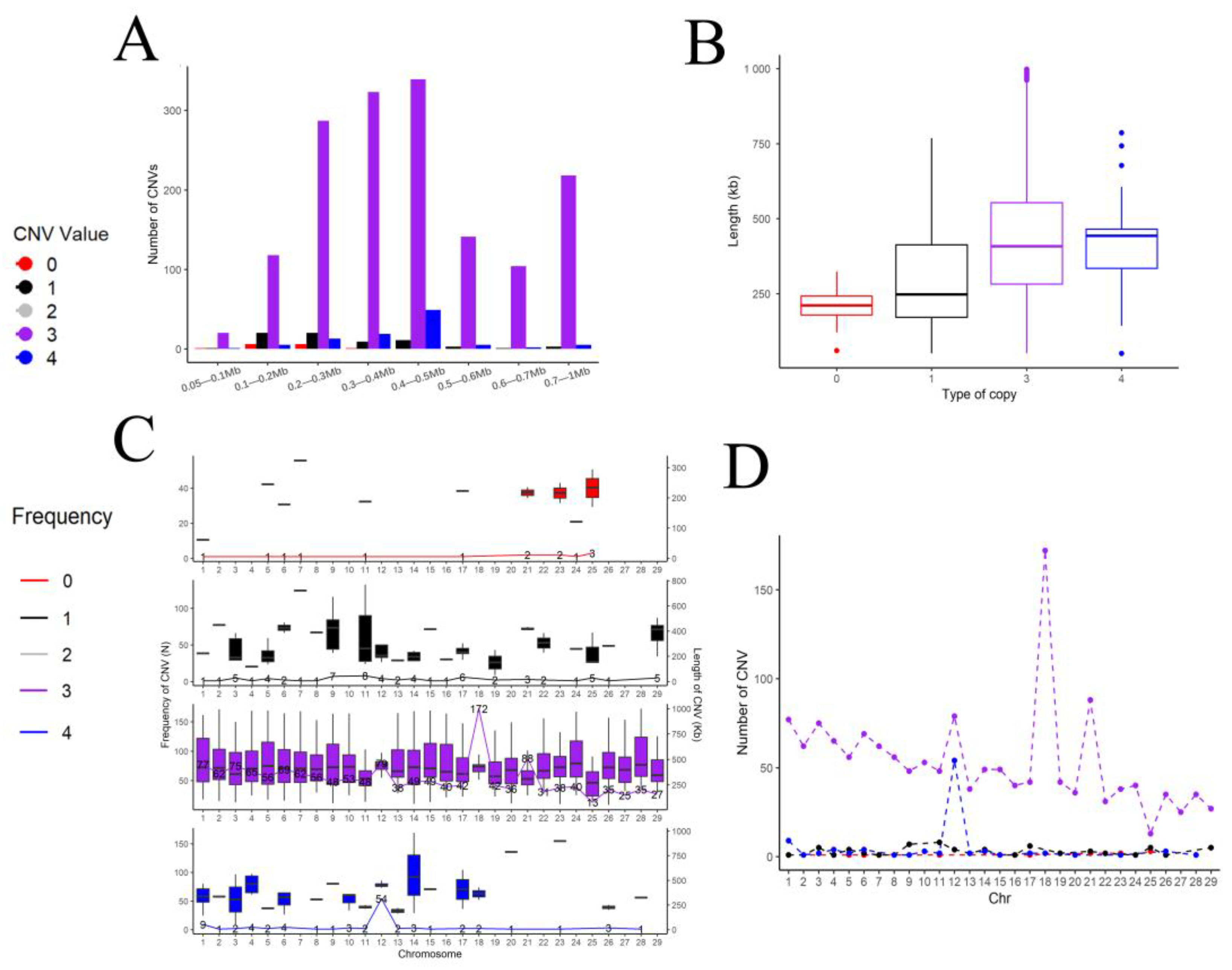
3.4. Genome Copy Number Variation in the Chinese Holstein Cattle in Jiangsu
3.5. Regional Distribution of Copy Number Variations in the Chinese Holstein Cattle in Jiangsu
3.6. Association Analysis of the CNVRs and Milk Production Traits of the Chinese Holstein Cattle in Jiangsu
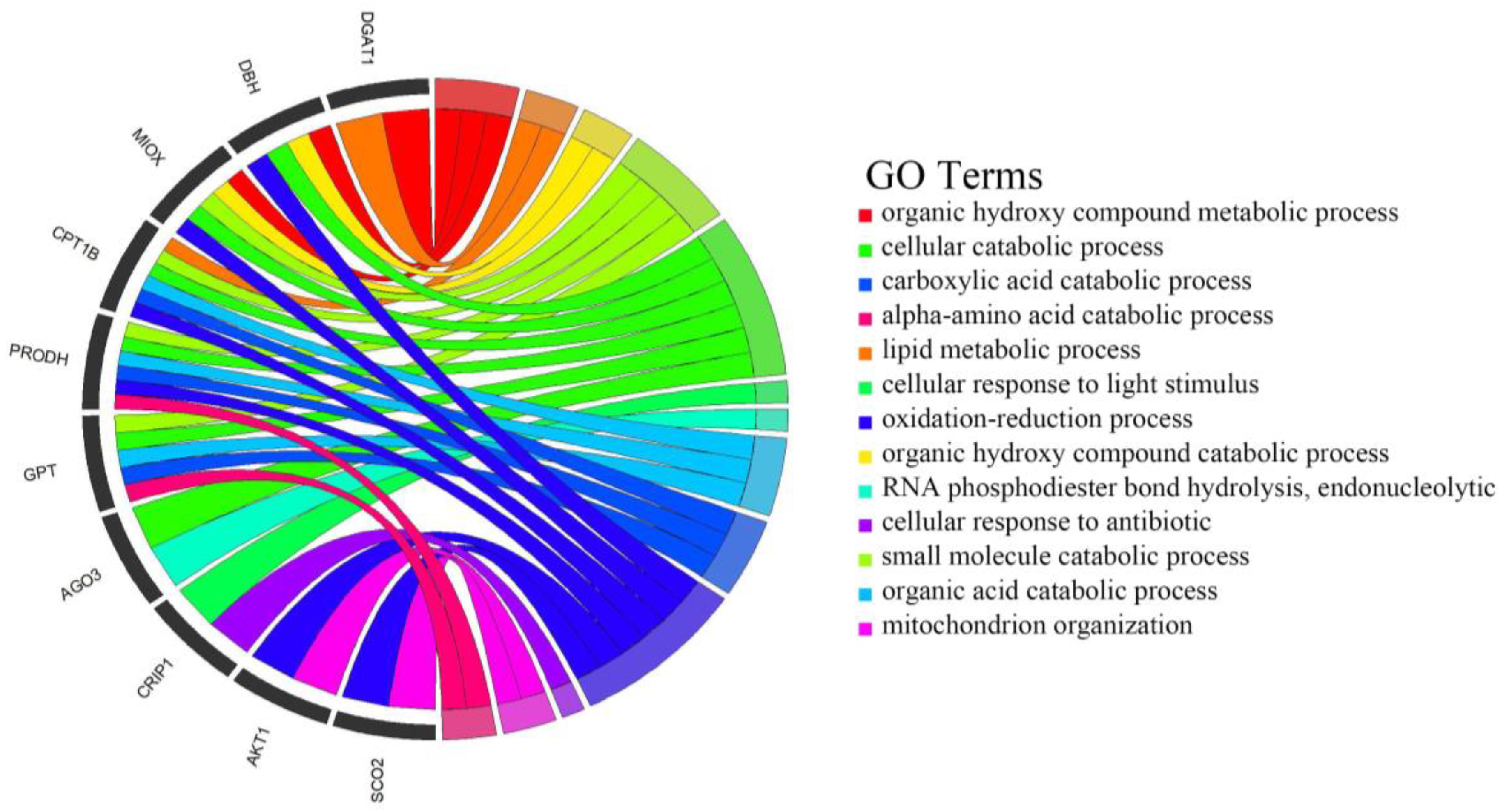
3.7. Enrichment Analysis of Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dallas, D.; Nielsen, S.D. Milk Peptidomics to Identify Functional Peptides and for Quality Control of Dairy Products. Methods Mol. Biol. 2018, 1719, 223–240. [Google Scholar] [PubMed]
- Nayeri, S.; Sargolzaei, M.; Abo-Ismail, M.K.; May, N.; Miller, S.P.; Schenkel, F.; Moore, S.S.; Stothard, P. Genome-wide association for milk production and female fertility traits in Canadian dairy Holstein cattle. BMC Genet. 2016, 17, 75. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, K.; Lagisz, M.; Nakagawa, S. Nonadditive genetic effects induce novel phenotypic distributions in male mating traits of F1 hybrids. Evolution 2021, 75, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Boichard, D.; Brochard, M. New phenotypes for new breeding goals in dairy cattle. Animal 2012, 6, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.F.; Bedere, N.; Douhard, F.; Oliveira, H.R.; Arnal, M.; Peñagaricano, F.; Schinckel, A.P.; Baes, C.F.; Miglior, F. Review: Genetic selection of high-yielding dairy cattle toward sustainable farming systems in a rapidly changing world. Animal 2021, 15 (Suppl. S1), 100292. [Google Scholar] [CrossRef]
- Seo, D.; Lee, D.H.; Jin, S.; Won, J.I.; Lim, D.; Park, M.; Kim, T.H.; Lee, H.K.; Kim, S.; Choi, I.; et al. Long-term artificial selection of Hanwoo (Korean) cattle left genetic signatures for the breeding traits and has altered the genomic structure. Sci. Rep. 2022, 12, 6438. [Google Scholar] [CrossRef]
- Xiang, R.; MacLeod, I.M.; Bolormaa, S.; Goddard, M.E. Genome-wide comparative analyses of correlated and uncorrelated phenotypes identify major pleiotropic variants in dairy cattle. Sci. Rep. 2017, 7, 9248. [Google Scholar] [CrossRef]
- van den Berg, I.; Boichard, D.; Lund, M.S. Comparing power and precision of within-breed and multibreed genome-wide association studies of production traits using whole-genome sequence data for 5 French and Danish dairy cattle breeds. J. Dairy Sci. 2016, 99, 8932–8945. [Google Scholar] [CrossRef]
- de Almeida Santana, M.H.; Junior, G.A.; Cesar, A.S.; Freua, M.C.; da Costa Gomes, R.; da Luz E Silva, S.; Leme, P.R.; Fukumasu, H.; Carvalho, M.E.; Ventura, R.V.; et al. Copy number variations and genome-wide associations reveal putative genes and metabolic pathways involved with the feed conversion ratio in beef cattle. J. Appl. Genet. 2016, 57, 495–504. [Google Scholar] [CrossRef]
- Jiang, L.; Jiang, J.; Yang, J.; Liu, X.; Wang, J.; Wang, H.; Ding, X.; Liu, J.; Zhang, Q. Genome-wide detection of copy number variations using high-density SNP genotyping platforms in Holsteins. BMC Genom. 2013, 14, 131. [Google Scholar] [CrossRef]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Lye, Z.N.; Purugganan, M.D. Copy Number Variation in Domestication. Trends Plant Sci. 2019, 24, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Macé, A.; Kutalik, Z.; Valsesia, A. Copy Number Variation. Methods Mol. Biol. 2018, 1793, 231–258. [Google Scholar] [PubMed]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef]
- Zhou, Y.; Utsunomiya, Y.T.; Xu, L.; el Hamidi Hay, A.; Bickhart, D.M.; Alexandre, P.A.; Rosen, B.D.; Schroeder, S.G.; Carvalheiro, R.; de Rezende Neves, H.H.; et al. Genome-wide CNV analysis reveals variants associated with growth traits in Bos indicus. BMC Genom. 2016, 17, 419. [Google Scholar] [CrossRef]
- Emerson, J.J.; Cardoso-Moreira, M.; Borevitz, J.O.; Long, M. Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science 2008, 320, 1629–1631. [Google Scholar] [CrossRef]
- Liu, G.E.; Ventura, M.; Cellamare, A.; Chen, L.; Cheng, Z.; Zhu, B.; Li, C.; Song, J.; Eichler, E.E. Analysis of recent segmental duplications in the bovine genome. BMC Genom. 2009, 10, 571. [Google Scholar] [CrossRef]
- Zhang, Z.; Peng, M.; Wen, Y.; Chai, Y.; Liang, J.; Yang, P.; Liu, X.; Li, J.; Huang, Y.; Li, L.; et al. Copy number variation of EIF4A2 loci related to phenotypic traits in Chinese cattle. Vet. Med. Sci. 2022, 8, 2147–2156. [Google Scholar] [CrossRef]
- Huddleston, J.; Chaisson, M.J.P.; Steinberg, K.M.; Warren, W.; Hoekzema, K.; Gordon, D.; Graves-Lindsay, T.A.; Munson, K.M.; Kronenberg, Z.N.; Vives, L.; et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2017, 27, 677–685. [Google Scholar] [CrossRef]
- Zhang, Z.; Hao, K. Using SAAS-CNV to Detect and Characterize Somatic Copy Number Alterations in Cancer Genomes from Next Generation Sequencing and SNP Array Data. Methods Mol. Biol. 2018, 1833, 29–47. [Google Scholar]
- Sahajpal, N.S.; Barseghyan, H.; Kolhe, R.; Hastie, A.; Chaubey, A. Optical Genome Mapping as a Next-Generation Cytogenomic Tool for Detection of Structural and Copy Number Variations for Prenatal Genomic Analyses. Genes 2021, 12, 398. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gao, Y.; Oswalt, A.; Fang, L.; Boschiero, C.; Neupane, M.; Sattler, C.G.; Li, C.J.; Seroussi, E.; Xu, L.; et al. Towards the detection of copy number variation from single sperm sequencing in cattle. BMC Genom. 2022, 23, 215. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.; Madsen, O.; Megens, H.J.; Frantz, L.A.; Bosse, M.; Crooijmans, R.P.; Groenen, M.A. Copy number variation in the speciation of pigs: A possible prominent role for olfactory receptors. BMC Genom. 2015, 16, 330. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.H.; Regitano, L.C.; Geistlinger, L.; Pértille, F.; Giachetto, P.F.; Brassaloti, R.A.; Morosini, N.S.; Zimmer, R.; Coutinho, L.L. Genome-Wide Detection of CNVs and Their Association with Meat Tenderness in Nelore Cattle. PLoS ONE 2016, 11, e0157711. [Google Scholar] [CrossRef] [PubMed]
- Pezer, Ž.; Harr, B.; Teschke, M.; Babiker, H.; Tautz, D. Divergence patterns of genic copy number variation in natural populations of the house mouse (Mus musculus domesticus) reveal three conserved genes with major population-specific expansions. Genome Res. 2015, 25, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Cheong, H.S.; Kim, L.H.; NamGung, S.; Park, T.J.; Chun, J.Y.; Kim, J.Y.; Pasaje, C.F.; Lee, J.S.; Shin, H.D. Identification of copy number variations and common deletion polymorphisms in cattle. BMC Genom. 2010, 11, 232. [Google Scholar] [CrossRef]
- Xu, L.; Cole, J.B.; Bickhart, D.M.; Hou, Y.; Song, J.; VanRaden, P.M.; Sonstegard, T.S.; Van Tassell, C.P.; Liu, G.E. Genome wide CNV analysis reveals additional variants associated with milk production traits in Holsteins. BMC Genom. 2014, 15, 683. [Google Scholar] [CrossRef]
- Lu, X.; Arbab, A.A.I.; Abdalla, I.M.; Liu, D.; Zhang, Z.; Xu, T.; Su, G.; Yang, Z. Genetic Parameter Estimation and Genome-Wide Association Study-Based Loci Identification of Milk-Related Traits in Chinese Holstein. Front. Genet. 2022, 12, 799664. [Google Scholar] [CrossRef]
- Li, J.; Gao, H.; Madsen, P.; Li, R.; Liu, W.; Bao, P.; Xue, G.; Gao, Y.; Di, X.; Su, G. Impact of the Order of Legendre Polynomials in Random Regression Model on Genetic Evaluation for Milk Yield in Dairy Cattle Population. Front. Genet. 2020, 11, 586155. [Google Scholar] [CrossRef]
- Nazar, M.; Lu, X.; Abdalla, I.M.; Ullah, N.; Fan, Y.; Chen, Z.; Arbab, A.A.I.; Mao, Y.; Yang, Z. Genome-Wide Association Study Candidate Genes on Mammary System-Related Teat-Shape Conformation Traits in Chinese Holstein Cattle. Genes 2021, 12, 2020. [Google Scholar] [CrossRef]
- Marees, A.T.; de Kluiver, H.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, e1608. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Cardone, M.F.; Wang, K.; Kim, E.S.; Matukumalli, L.K.; Ventura, M.; Song, J.; VanRaden, P.M.; et al. Genomic characteristics of cattle copy number variations. BMC Genom. 2011, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Li, M.; Liu, M.; Liu, S.; Pan, M.G.; Wiggans, G.R.; Rosen, B.D.; Liu, G.E. Copy number variation analysis reveals variants associated with milk production traits in dairy goats. Genomics 2020, 112, 4934–4937. [Google Scholar] [CrossRef] [PubMed]
- Duchemin, S.I.; Nilsson, K.; Fikse, W.F.; Stålhammar, H.; Buhelt Johansen, L.; Stenholdt Hansen, M.; Lindmark-Månsson, H.; de Koning, D.J.; Paulsson, M.; Glantz, M. Genetic parameters for noncoagulating milk, milk coagulation properties, and detailed milk composition in Swedish Red Dairy Cattle. J. Dairy Sci. 2020, 103, 8330–8342. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Hu, H.; Zhang, J.; Ma, Y.; Han, L.; Hao, F.; Jiang, Y.; Ma, Y. Estimation of Genetic Parameters for Conformation Traits and Milk Production Traits in Chinese Holsteins. Animals 2022, 13, 100. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Matukumalli, L.K.; Li, C.; Song, J.; Gasbarre, L.C.; Van Tassell, C.P.; Sonstegard, T.S. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. Funct. Integr. Genom. 2012, 12, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Xi, H.; Chen, J.; Peng, Y.; Jia, Z.; Yang, S.; Hu, J.; Pang, J.; Zhang, Y.; Hu, R.; et al. Integrated CNV-seq, karyotyping and SNP-array analyses for effective prenatal diagnosis of chromosomal mosaicism. BMC Med Genom. 2021, 14, 56. [Google Scholar] [CrossRef]
- de Araújo Lima, L.; Wang, K. PennCNV in whole-genome sequencing data. BMC Bioinform. 2017, 18 (Suppl. S11), 383. [Google Scholar] [CrossRef]
- Marenne, G.; Rodríguez-Santiago, B.; Closas, M.G.; Pérez-Jurado, L.; Rothman, N.; Rico, D.; Pita, G.; Pisano, D.G.; Kogevinas, M.; Silverman, D.T.; et al. Assessment of copy number variation using the Illumina Infinium 1M SNP-array: A comparison of methodological approaches in the Spanish Bladder Cancer/EPICURO study. Hum. Mutat. 2011, 32, 240–248. [Google Scholar] [CrossRef]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Månér, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Hou, Y.; Schroeder, S.G.; Alkan, C.; Cardone, M.F.; Matukumalli, L.K.; Song, J.; Schnabel, R.D.; Ventura, M.; Taylor, J.F.; et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012, 22, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Jiang, J.; Wang, J.; Ding, X.; Liu, J.; Zhang, Q. Genome-wide identification of copy number variations in Chinese Holstein. PLoS ONE 2012, 7, e48732. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.C.; da Silva, V.H.; Goes, C.P.; Moreira, G.C.M.; Godoy, T.F.; Ibelli, A.M.G.; Peixoto, J.O.; Cantão, M.E.; Ledur, M.C.; de Rezende, F.M.; et al. Genome-wide detection of CNVs and their association with performance traits in broilers. BMC Genom. 2021, 22, 354. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.E.; Bickhart, D.M. Copy number variation in the cattle genome. Funct. Integr. Genom. 2012, 12, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.H.; Ben-Dor, A.; Tsalenko, A.; Sampas, N.; Rodriguez-Revenga, L.; Tran, C.W.; Scheffer, A.; Steinfeld, I.; Tsang, P.; Yamada, N.A.; et al. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008, 82, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Jiang, J.; Yang, S.; Hou, Y.; Liu, G.E.; Zhang, S.; Zhang, Q.; Sun, D. CNV discovery for milk composition traits in dairy cattle using whole genome resequencing. BMC Genom. 2017, 18, 265. [Google Scholar] [CrossRef]
- Nafikov, R.A.; Schoonmaker, J.P.; Korn, K.T.; Noack, K.; Garrick, D.J.; Koehler, K.J.; Minick-Bormann, J.; Reecy, J.M.; Spurlock, D.E.; Beitz, D.C. Polymorphisms in lipogenic genes and milk fatty acid composition in Holstein dairy cattle. Genomics 2014, 104 Pt 6, 572–581. [Google Scholar] [CrossRef]
- Osorio, J.S.; Lohakare, J.; Bionaz, M. Biosynthesis of milk fat, protein, and lactose: Roles of transcriptional and posttranscriptional regulation. Physiol. Genom. 2016, 48, 231–256. [Google Scholar] [CrossRef]
- Bovenhuis, H.; Visker, M.H.P.W.; Poulsen, N.A.; Sehested, J.; van Valenberg, H.J.F.; van Arendonk, J.A.M.; Larsen, L.B.; Buitenhuis, A.J. Effects of the diacylglycerol o-acyltransferase 1 (DGAT1) K232A polymorphism on fatty acid, protein, and mineral composition of dairy cattle milk. J. Dairy Sci. 2016, 99, 3113–3123. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.W.; Ma, J.; Yang, C.L.; Chen, L.L.; He, Q.Y.; Coleman, D.N.; Wang, T.Z.; Jiang, X.L.; Luo, J.; Ma, Y.; et al. Phosphatase and tensin homolog (PTEN) suppresses triacylglycerol accumulation and monounsaturated fatty acid synthesis in goat mammary epithelial cells. J. Dairy Sci. 2021, 104, 7283–7294. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhou, J.; Chen, C.J.; Zhang, J.; Wen, W.; Tian, J.; Zhang, Z.; Gu, Y. GWAS-Based Identification of New Loci for Milk Yield, Fat, and Protein in Holstein Cattle. Animals 2020, 10, 2048. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.R.; Cant, J.P.; Brito, L.F.; Feitosa, F.L.B.; Chud, T.C.S.; Fonseca, P.A.S.; Jamrozik, J.; Silva, F.F.; Lourenco, D.A.L.; Schenkel, F.S. Genome-wide association for milk production traits and somatic cell score in different lactation stages of Ayrshire, Holstein, and Jersey dairy cattle. J. Dairy Sci. 2019, 102, 8159–8174. [Google Scholar] [CrossRef]
- Sanchez, M.P.; Ramayo-Caldas, Y.; Wolf, V.; Laithier, C.; El Jabri, M.; Michenet, A.; Boussaha, M.; Taussat, S.; Fritz, S.; Delacroix-Buchet, A.; et al. Sequence-based GWAS, network and pathway analyses reveal genes co-associated with milk cheese-making properties and milk composition in Montbéliarde cows. Genet. Sel. Evol. 2019, 51, 34. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Parity | NI | NR | Mean | SD | Median | Min | Max | Skew | Kurtosis |
|---|---|---|---|---|---|---|---|---|---|---|
| MY (kg) | 1 | 6437 | 46,749 | 30.39 | 7.98 | 30.70 | 5.00 | 75.00 | −0.08 | 0.25 |
| 2 | 3402 | 24,864 | 36.28 | 10.79 | 36.00 | 5.00 | 80.00 | 0.12 | 0.17 | |
| 3 | 3124 | 23,162 | 36.94 | 10.95 | 36.50 | 5.00 | 79.00 | 0.10 | −0.10 | |
| 1–3 | 9834 | 95,375 | 33.33 | 6.23 | 33.00 | 5.00 | 80.00 | 0.31 | 0.43 | |
| MFR (%) | 1 | 6263 | 45,814 | 3.84 | 0.96 | 3.79 | 0.33 | 9.99 | 0.48 | 1.55 |
| 2 | 3196 | 23,785 | 3.91 | 1.02 | 3.87 | 0.32 | 9.99 | 0.50 | 1.34 | |
| 3 | 3088 | 23,104 | 3.97 | 1.04 | 3.92 | 0.36 | 9.57 | 0.55 | 1.23 | |
| 1–3 | 9497 | 92,763 | 3.88 | 1.00 | 3.82 | 0.30 | 9.99 | 0.52 | 1.46 | |
| MFY (kg) | 1 | 6263 | 45,814 | 1.17 | 0.40 | 1.14 | 0.06 | 4.58 | 0.70 | 1.62 |
| 2 | 3196 | 23,785 | 1.41 | 0.53 | 1.35 | 0.08 | 4.90 | 0.77 | 1.41 | |
| 3 | 3088 | 23,104 | 1.46 | 0.55 | 1.40 | 0.08 | 5.74 | 0.77 | 1.47 | |
| 1–3 | 9497 | 92,763 | 1.29 | 0.50 | 1.23 | 0.60 | 5.74 | 0.93 | 2.00 | |
| MPR (%) | 1 | 6263 | 45,814 | 3.27 | 0.36 | 3.25 | 0.30 | 9.46 | 0.74 | 6.50 |
| 2 | 3196 | 23,785 | 3.29 | 0.38 | 3.26 | 0.75 | 7.27 | 0.71 | 3.43 | |
| 3 | 3088 | 23,104 | 3.30 | 0.39 | 3.27 | 1.22 | 7.22 | 0.62 | 2.94 | |
| 1–3 | 9497 | 92,763 | 3.28 | 0.37 | 3.25 | 0.30 | 9.46 | 0.75 | 4.95 | |
| MPY (kg) | 1 | 6263 | 45,814 | 1.00 | 0.25 | 1.00 | 0.11 | 2.43 | −0.01 | 0.59 |
| 2 | 3196 | 23,785 | 1.18 | 0.34 | 1.18 | 0.17 | 3.27 | 0.22 | 0.83 | |
| 3 | 3088 | 23,104 | 1.21 | 0.33 | 1.2 | 0.13 | 2.76 | 0.07 | 0.26 | |
| 1–3 | 9497 | 92,763 | 1.09 | 0.32 | 1.07 | 0.10 | 4.58 | 0.36 | 1.02 |
| Traits | Parity | Parity 1 | Parity 2 | Parity 3 |
|---|---|---|---|---|
| MY | 1 | 1.000 | 0.478 ** | 0.512 ** |
| 2 | 0.596 (0.098) | 1.000 | 0.453 ** | |
| 3 | 0.857 (0.088) | 0.867 (0.102) | 1.000 | |
| MFR | 1 | 1.000 | 0.380 ** | 0.421 ** |
| 2 | 0.913 (0.078) | 1.000 | 0.419 ** | |
| 3 | 0.963 (0.141) | 0.989 (0.143) | 1.000 | |
| MFY | 1 | 1.000 | 0.362 ** | 0.358 ** |
| 2 | 0.623 (0.158) | 1.000 | 0.324 ** | |
| 3 | 0.779 (0.171) | 0.975 (0.205) | 1.000 | |
| MPR | 1 | 1.000 | 0.444 ** | 0.554 ** |
| 2 | 0.902 (0.054) | 1.000 | 0.546 ** | |
| 3 | 0.963 (0.053) | 0.985 (0.078) | 1.000 | |
| MPY | 1 | 1.000 | 0.420 ** | 0.436 ** |
| 2 | 0.552 (0.117) | 1.000 | 0.399 ** | |
| 3 | 0.858 (0.122) | 0.815 (0.144) | 1.000 |
| CNV Type | Counts | Length | Min-Length | Max-Length |
|---|---|---|---|---|
| 0 | 14 | 207,115 | 60,725 | 323,251 |
| 1 | 68 | 302,899 | 50,880 | 768,584 |
| 3 | 1550 | 440,638 | 50,683 | 998,290 |
| 4 | 99 | 414,047 | 50,735 | 980,071 |
| Traits | CNVR ID | Chr | Start | End | Size | Description | Adjusted p-Value |
|---|---|---|---|---|---|---|---|
| MY | CNVR_155 | 17 | 71,644,239 | 71,885,069 | 240,831 | Loss | 0.031 |
| CNVR_156 | 17 | 72,803,799 | 73,155,293 | 351,495 | Loss | 0.033 | |
| CNVR_106 | 11 | 103,230,152 | 104,256,742 | 1,026,591 | Mixed | 0.040 | |
| MFR | CNVR_124 | 14 | 146,715 | 891,340 | 744,626 | Mixed | 0.000 |
| CNVR_140 | 15 | 69,494,372 | 70,392,722 | 898,351 | Gain | 0.011 | |
| CNVR_184 | 21 | 69,165,607 | 69,778,711 | 613,105 | Loss | 0.012 | |
| CNVR_32 | 3 | 63,079,015 | 65,566,990 | 2,487,976 | Gain | 0.015 | |
| MFY | CNVR_148 | 17 | 22,459,660 | 23,169,353 | 709,694 | Gain | 0.01 |
| CNVR_106 | 11 | 103,230,152 | 104,256,742 | 1,026,591 | Mixed | 0.018 | |
| CNVR_113 | 12 | 58,857,555 | 60,002,395 | 1,144,841 | Gain | 0.021 | |
| CNVR_124 | 14 | 146,715 | 891,340 | 744,626 | Mixed | 0.028 | |
| CNVR_156 | 17 | 72,803,799 | 73,155,293 | 351,495 | Loss | 0.030 | |
| CNVR_161 | 18 | 48,610,254 | 48,869,465 | 1,026,591 | Gain | 0.047 | |
| MPR | CNVR_106 | 11 | 103,230,152 | 104,256,742 | 1,026,591 | Mixed | 0.013 |
| CNVR_155 | 17 | 71,644,239 | 71,885,069 | 240,831 | Loss | 0.020 | |
| CNVR_156 | 17 | 72,803,799 | 73,155,293 | 351,495 | Loss | 0.035 | |
| CNVR_107 | 11 | 104,418,358 | 106,541,521 | 2,123,164 | Mixed | 0.037 | |
| CNVR_59 | 5 | 119,074,417 | 119,998,002 | 923,586 | Mixed | 0.042 | |
| CNVR_40 | 3 | 109,548,750 | 110,571,030 | 1,022,281 | Mixed | 0.048 |
| Pathway | Description | Gene Name | FDR(q)-p-Values |
|---|---|---|---|
| bta04920 | Adipocytokine signaling pathway | AKT1, NFKBIB, RXRA, TRAF2, CPT1B | 0.0003 |
| bta01522 | Endocrine resistance | NOTCH1, AKT1,JAG2, MAPK12, MAPK11 | 0.001 |
| bta04658 | Th1 and Th2 cell differentiation | NOTCH1, JAG2, NFKBIB, MAPK12, MAPK11 | 0.0013 |
| bta04664 | Fc epsilon RI signaling pathway | AKT1, VAV2, MAPK12, MAPK11 | 0.0026 |
| bta04621 | NOD-like receptor signaling pathway | CARD9, SHARPIN, NFKBIB, TRAF2, MAPK12, MAPK11 | 0.0051 |
| bta00513 | Various types of N-glycan biosynthesis | MAN1B1, MAN1B1, ALG12 | 0.0052 |
| bta00510 | N-Glycan biosynthesis | MAN1B1, MAN1B1, ALG12 | 0.0088 |
| bta00564 | Glycerophospholipid metabolism | AGPAT2, PLD4, CHKB | 0.0104 |
| bta04625 | C-type lectin receptor signaling pathway | CARD9, AKT1, MAPK12, MAPK11 | 0.0107 |
| bta04622 | RIG-I-like receptor signaling pathway | NFKBIB, TRAF2, MAPK12, MAPK11 | 0.0111 |
| bta04370 | VEGF signaling pathway | AKT1, MAPK12, MAPK11 | 0.0119 |
| bta04659 | Th17 cell differentiation | NFKBIB, RXRA, MAPK12, MAPK11 | 0.015 |
| bta04071 | Sphingolipid signaling pathway | AKT1, TRAF2, MAPK12, MAPK11 | 0.0168 |
| bta04722 | Neurotrophin signaling pathway | AKT1, NFKBIB, MAPK12, MAPK11 | 0.0178 |
| bta00601 | Glycosphingolipid biosynthesis—lacto and neolacto series | ABO, FUT7 | 0.0249 |
| bta04917 | Prolactin signaling pathway | AKT1, MAPK12, MAPK11 | 0.0315 |
| bta04015 | Rap1 signaling pathway | AKT1, VAV2, GRIN1, MAPK12, MAPK11 | 0.0326 |
| bta04914 | Progesterone-mediated oocyte maturation | AKT1, MAPK12, MAPK11 | 0.0376 |
| bta00250 | Alanine, aspartate and glutamate metabolism | GPT, ADSS1 | 0.0387 |
| bta00350 | Tyrosine metabolism | COMT, DBH | 0.0405 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Lu, X.; Jiang, H.; Yang, Z.; Xu, T. Descriptive Statistics and Genome-Wide Copy Number Analysis of Milk Production Traits of Jiangsu Chinese Holstein Cows. Animals 2024, 14, 17. https://doi.org/10.3390/ani14010017
Zhu H, Lu X, Jiang H, Yang Z, Xu T. Descriptive Statistics and Genome-Wide Copy Number Analysis of Milk Production Traits of Jiangsu Chinese Holstein Cows. Animals. 2024; 14(1):17. https://doi.org/10.3390/ani14010017
Chicago/Turabian StyleZhu, Hao, Xubin Lu, Hui Jiang, Zhangping Yang, and Tianle Xu. 2024. "Descriptive Statistics and Genome-Wide Copy Number Analysis of Milk Production Traits of Jiangsu Chinese Holstein Cows" Animals 14, no. 1: 17. https://doi.org/10.3390/ani14010017