
Analysis of Genetic Diversity and Population Structure of Endemic Endangered Goose (Anser cygnoides) Breeds Based on Mitochondrial CYTB

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
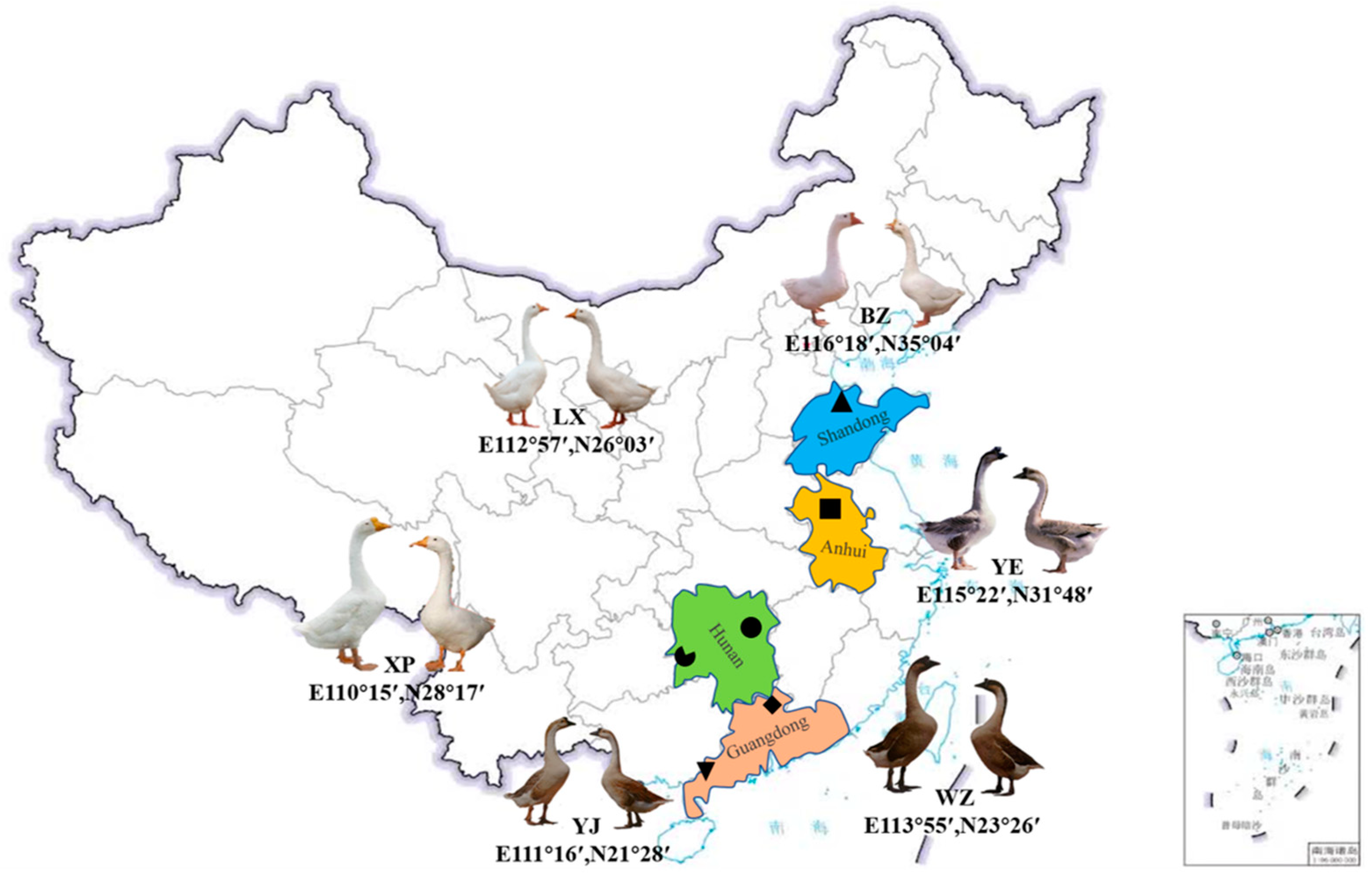
2.2. Experimental Design and Facilities
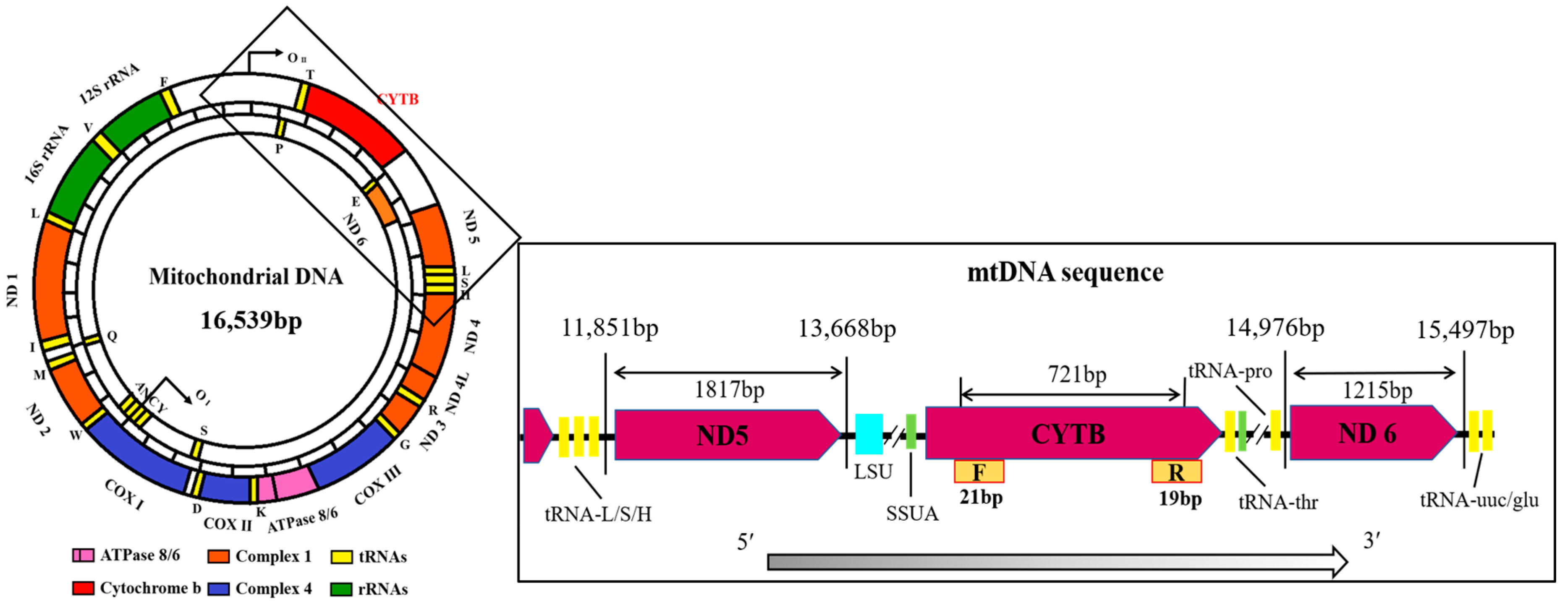
2.3. Sequence Analysis of Genetic Diversity
2.4. Haplotype Network and Population Structure
3. Results
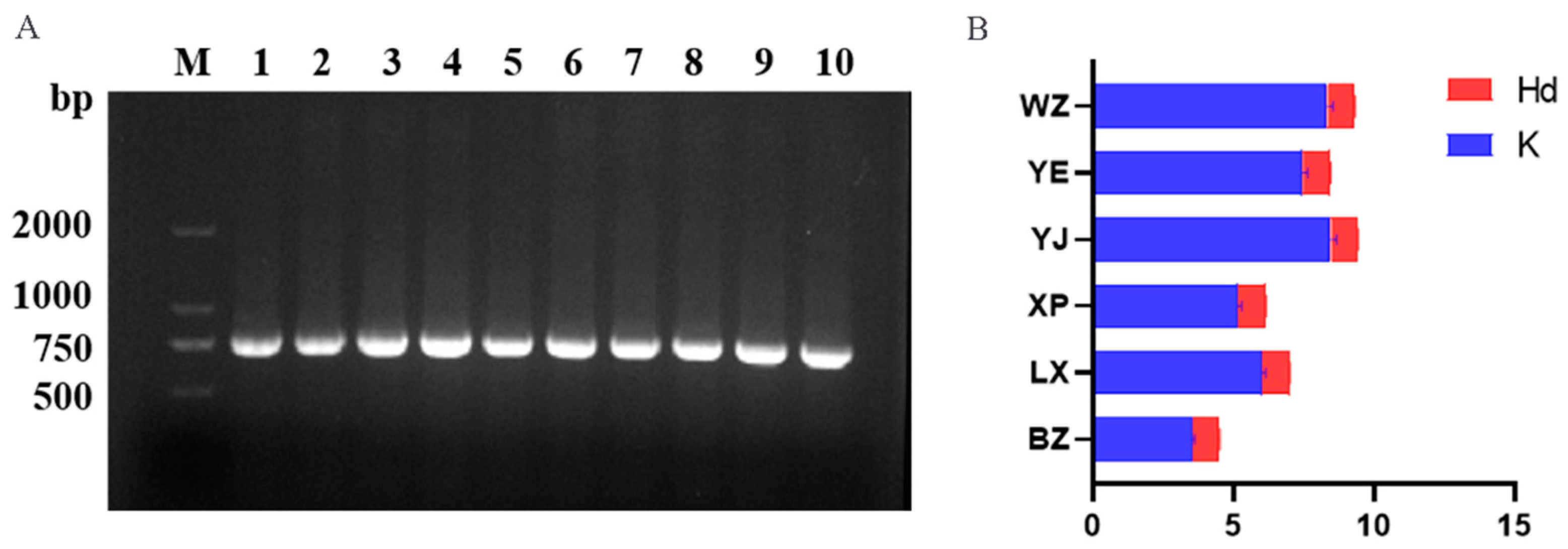
3.1. CYTB Gene Sequence Amplification and Verification
3.2. Analysis of Genetic Structure of Endemic Endangered Goose Breeds
3.2.1. CYTB Gene Locus Information and Nucleic Acid Diversity Analysis
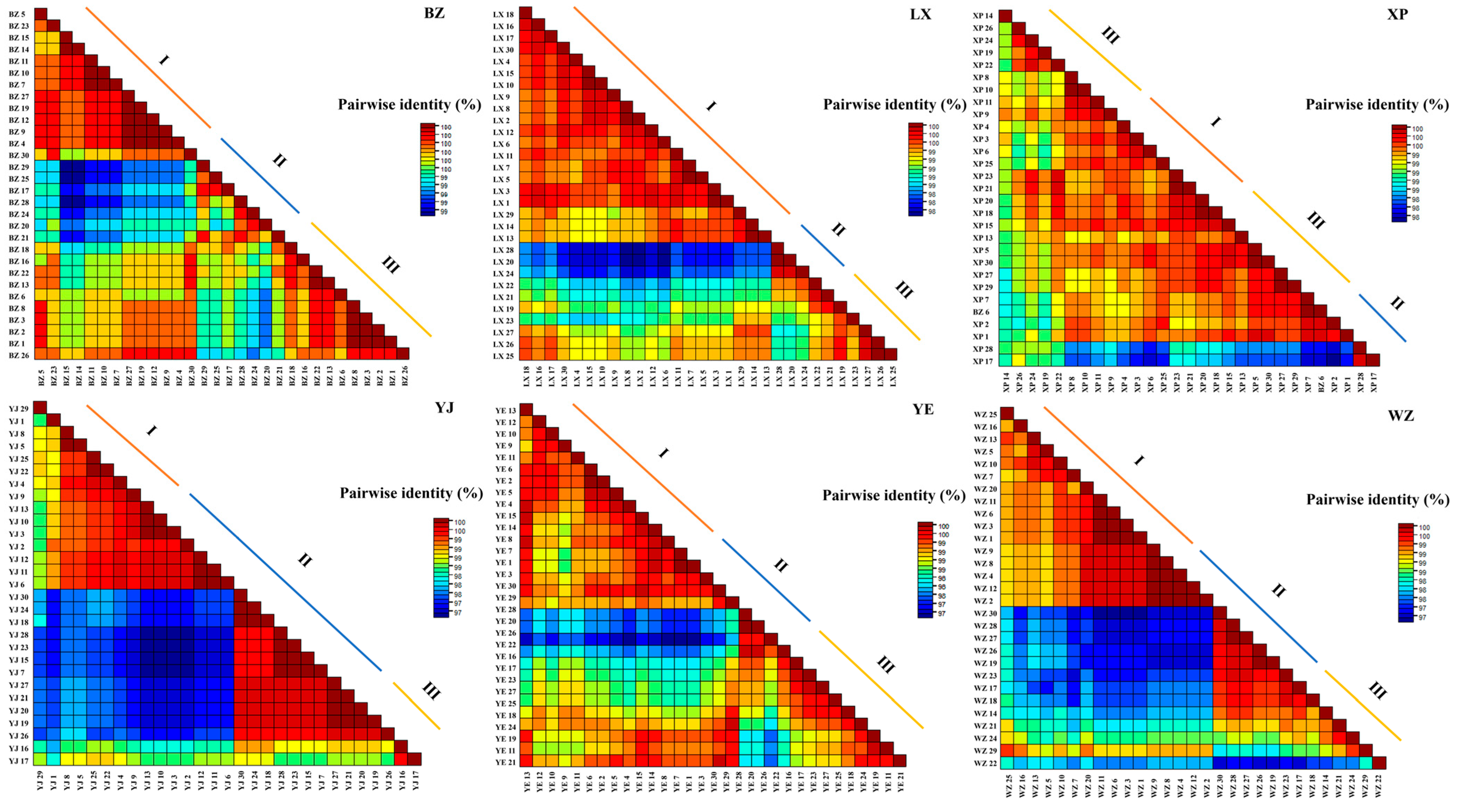
3.2.2. Comparison of Base Homology within CYTB Gene Sequence of Each Breed
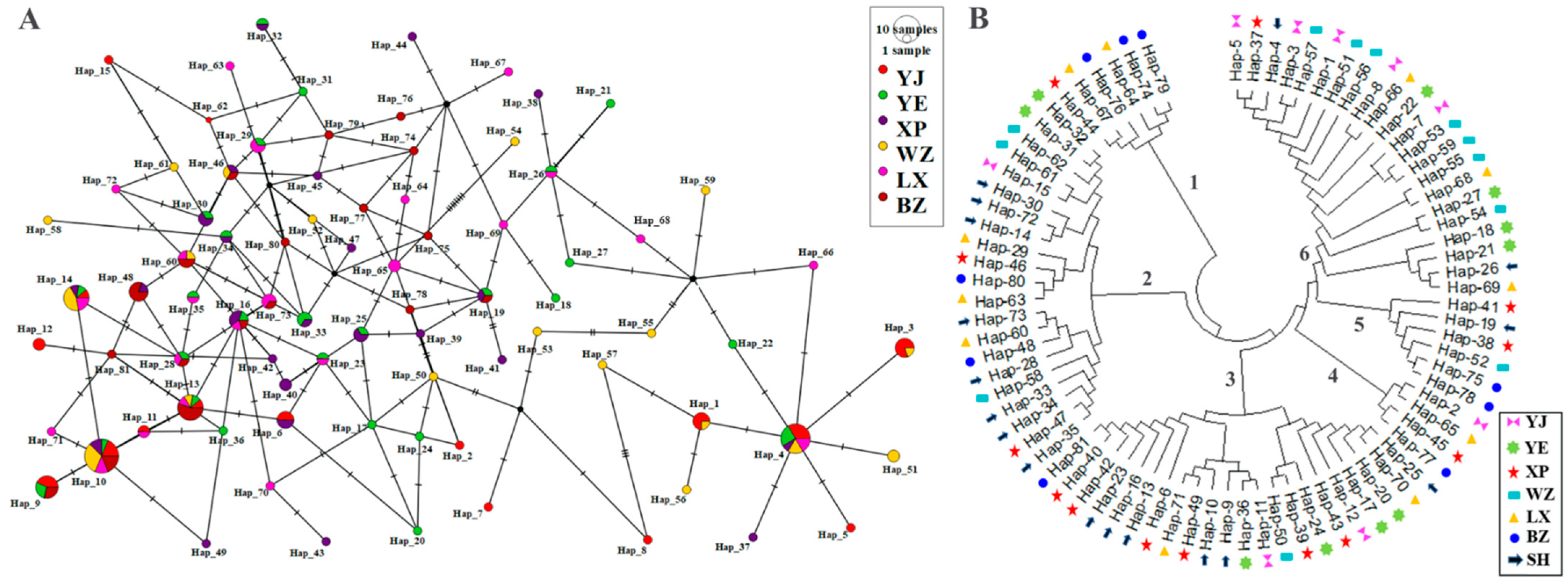
3.2.3. MtDNA CYTB Gene Haplotype Distribution and Proportion Analysis
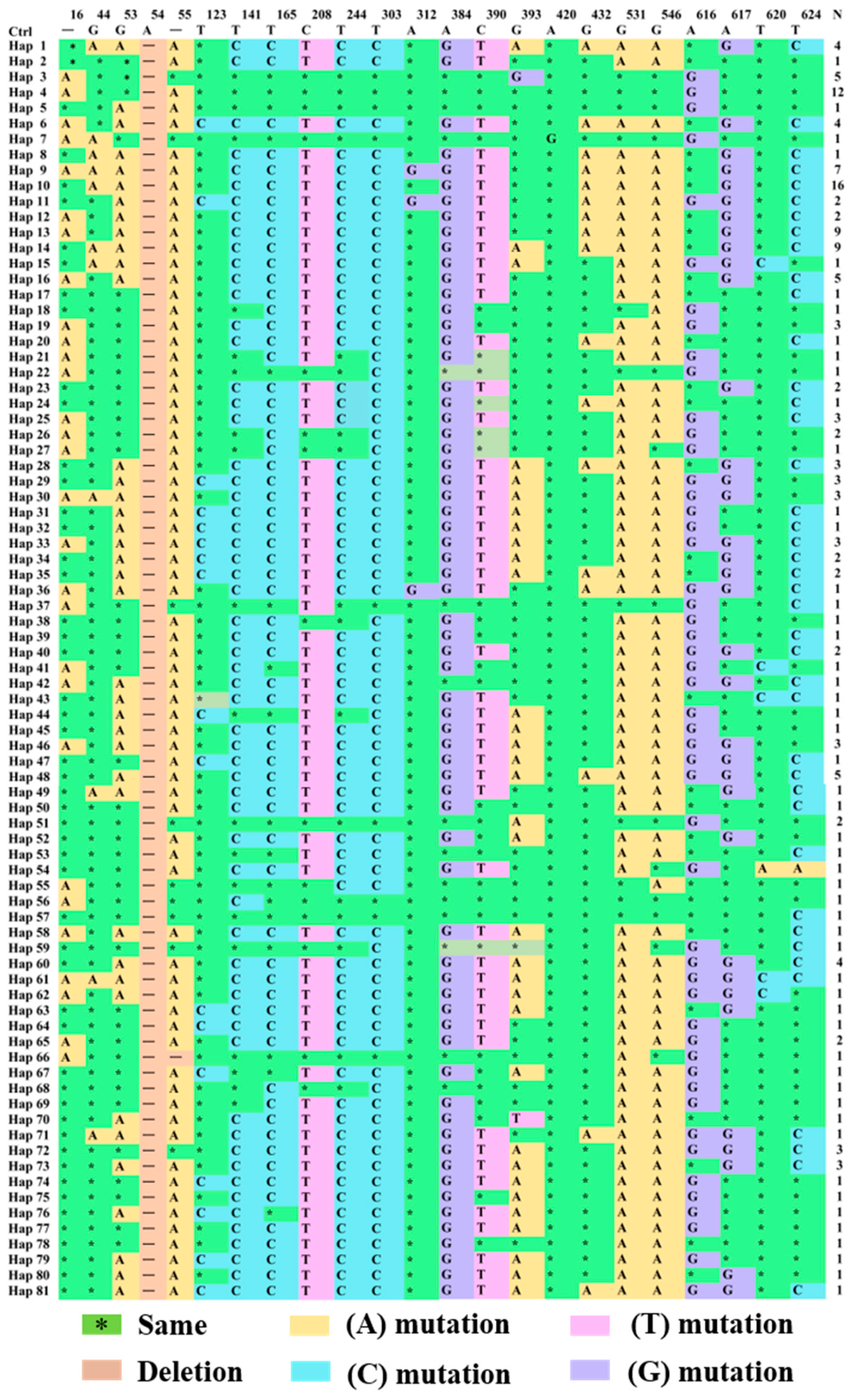
3.2.4. Analysis of Variable Loci of CYTB Gene in Endemic Endangered Geese
3.3. Analysis of Clustering Relationships of CYTB Genes in Endemic Endangered Geese
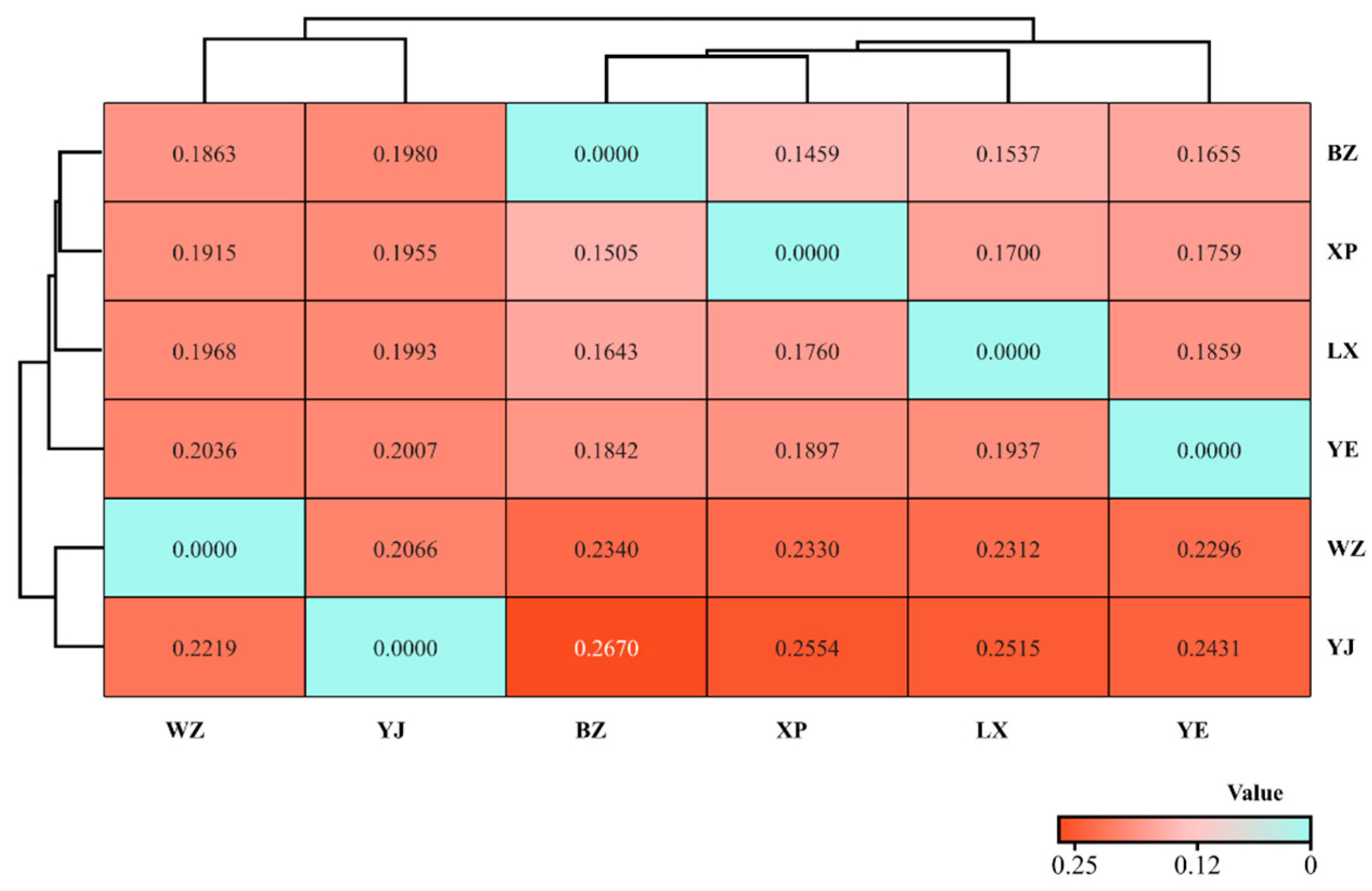
3.4. Genetic Differentiation Index and Gene Flow of Endemic Endangered Breeds of Geese
3.5. Population Historical Dynamic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruegg, K.; Turbek, S. Estimating global genetic diversity loss. Science 2022, 23, 1384–1385. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Gao, X.; Zeng, G.; Hua, S.; Zhong, M.; Li, X.; Li, X. Coupling Modern Portfolio Theory and Marxan enhances the efficiency of Lesser White-fronted Goose’s (Anser erythropus) habitat conservation. Sci. Rep. 2018, 9, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Ma, L.; Yi, Y.; Lin, T. Assessment of heavy metal pollution and exposure risk for migratory birds—A case study of Caohai wetland in Guizhou Plateau (China). Environ. Pollut. 2021, 15, 116564. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zheng, S.; Huang, M.; Tang, H.; Qiu, X.; Chen, S.; Wang, Z.; Zhou, Z.; Gao, Y.; Xiong, Y.; et al. Chromosome-level genome and population genomics reveal evolutionary characteristics and conservation status of Chinese indigenous geese. Commun. Biol. 2022, 7, 1191. [Google Scholar] [CrossRef] [PubMed]
- Weng, K.; Huo, W.; Zhang, Y.; Zhang, Y.; Chen, G.; Xu, Q. Principal component analysis of body size, reproductive performance, and ecological characteristics of local goose breeds in China. J. Sichuan Agric. Univ. 2020, 38, 225–233. [Google Scholar]
- Chen, G.; Liu, J.; Xu, Q. Current Status and Prospects of Conservation and Utilization of Goose Genetic Resources in China. China Poult. Ind. Guide 2022, 39, 6–12. [Google Scholar]
- Pumpitakkul, V.; Chetruengchai, W.; Srichomthong, C.; Phokaew, C.; Pootakham, W.; Sonthirod, C.; Nawae, W.; Tongsima, S.; Wangkumhang, P.; Wilantho, A.; et al. Comparative genomics and genome-wide SNPs of endangered Eld’s deer provide breeder selection for inbreeding avoidance. Sci. Rep. 2023, 13, 19806–19815. [Google Scholar] [CrossRef] [PubMed]
- Andres, K.; Kapkowska, E. Applicability of anatid and galliform microsatellite markers to the genetic diversity studies of domestic geese (Anser anser domesticus) through the genotyping of the endangered zatorska breed. BMC Res. Notes 2011, 16, 60–65. [Google Scholar] [CrossRef]
- Ni, H.; Zhang, Y.; Yang, Y.; Yin, Y.; Xie, H.; Zheng, J.; Dong, L.; Diao, J.; Wei, M.; Lv, Z.; et al. Whole genome sequencing of a wild swan goose breeds. Front. Genet. 2023, 24, 1038606. [Google Scholar] [CrossRef]
- Lai, F.Y.; Tu, P.A.; Ding, S.T.; Lin, M.J.; Chang, S.C.; Lin, E.C.; Lo, L.L.; Wang, P.H. Survey of genetic structure of geese using novel microsatellite markers. Asian-Australas J. Anim. Sci. 2018, 31, 167–179. [Google Scholar] [CrossRef]
- Wen, J.; Li, H.; Wang, H.; Yu, J.; Zhu, T.; Zhang, J.; Li, X.; Jiang, Z.; Ning, Z.; Qu, L. Origins, timing and introgression of domestic geese revealed by whole genome data. J. Anim. Sci. Biotechnol. 2023, 13, 251426. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, M.E.; Ruokonen, M.; Alexander, M.; Aspi, J.; Pyhäjärvi, T.; Searle, J.B. Relationship between wild greylag and European domestic geese based on mitochondrial DNA. Anim. Genet. 2015, 46, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Weiß, B.M.; Poggemann, K.; Olek, K.; Foerster, K.; Hirschenhauser, K. Isolation and characterization of microsatellite marker loci in the greylag goose (Anser anser). Mol. Ecol. Resour. 2008, 8, 1411–1413. [Google Scholar] [CrossRef]
- Liu, D.; Zhou, Y.; Fei, Y.; Xie, C.; Hou, S. Mitochondrial genome of the critically endangered Baer’s Pochard, Aythya baeri, and its phylogenetic relationship with other Anatidae species. Sci. Rep. 2021, 21, 24302. [Google Scholar] [CrossRef]
- Cooley, A.M. Mitochondrial DNA Analysis. Methods Mol. Biol. 2023, 2685, 331–349. [Google Scholar]
- Taillebois, L.; Castelin, M.; Ovenden, J.R.; Bonillo, C.; Keith, P. Contrasting genetic structure among breeds of two amphidromous fish species (Sicydiinae) in the Central West Pacific. PLoS ONE 2013, 9, e75465. [Google Scholar]
- Zardoya, R.; Meyer, A. Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Mol. Biol. Evol. 1996, 13, 933–942. [Google Scholar] [CrossRef]
- Abdel-Kafy, E.M.; Ramadan, S.I.; Ali, W.H.; Youssef, S.F.; Shabaan, H.A.; El-Deighadi, A.; Inoue-Murayama, M. Genetic and Phenotypic Characterization of Domestic Geese (Anser anser) in Egypt. Animals 2021, 11, 3106. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.X.; Tang, X.; Lu, J.X.; Fang, Y.F.; Gu, R.; Ma, Y.P.; Huang, S.H.; Ge, Q.L.; Gao, Y.S. Genetic diversity and phylogenetic analysis of mitochondrial ND6 gene in five black bone chicken breeds. Northwest Agric. J. 2023, 32, 1159–1165. [Google Scholar]
- Zhang, Y.; Wang, M.Q.; Mu, C.Y. Sequence Analysis of COⅠ Barcode and Upstream tRNA of Goose Mitochondria. Acta-Agric. Zhejiangensis 2017, 29, 51–57. (In Chinese) [Google Scholar]
- Zhang, Y.; Manzoor, A.; Wang, X. Mitochondrial DNA analysis reveals spatial genetic structure and high genetic diversity of Massicus raddei (Blessig) (Coleoptera: Cerambycidae) in China. Ecol. Evol. 2020, 10, 11657–11670. [Google Scholar] [CrossRef]
- Guo, Q.; Zhu, Q.D.; Zhou, Z.J.; Shi, F.M. Phylogeography and Genetic Structure of the Bush Cricket Decma fissa (Orthoptera, Tettigoniidae) in Southern China. Zool Stud. 2023, 12, 32–40. [Google Scholar]
- Gonçalves, V.F. Mitochondrial Genetics. Adv. Exp. Med. Biol. 2019, 1158, 247–255. [Google Scholar] [PubMed]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta. 1999, 9, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Filloux, D.; Fernandez, E.; Comstock, J.C.; Mollov, D.; Roumagnac, P.; Rott, P. Viral Metagenomic-Based Screening of Sugarcane from Florida Reveals Occurrence of Six Sugarcane-Infecting Viruses and High Prevalence of Sugarcane yellow leaf virus. Plant Dis. 2018, 102, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 16, 152008. [Google Scholar] [CrossRef]
- Leigh, J.; Bryant, D. PopART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Laxmivandana, R.; Vashi, Y.; Kalita, D.; Banik, S.; Sahoo, N.R.; Naskar, S. Genetic diversity in mitochondrial DNA D-loop region of indigenous pig breeds of India. J. Genet. 2022, 101, 5–11. [Google Scholar] [CrossRef]
- Zhang, Q.; Tyler-Smith, C.; Long, Q. An extended Tajima’s D neutrality test incorporating SNP calling and imputation uncertainties. Stat. Interface 2015, 8, 447–456. [Google Scholar] [CrossRef]
- Wang, S.H.; Zhang, C.; Shang, M.; Wu, X.G.; Cheng, Y.X. Genetic diversity and population structure of native mitten crab (Eriocheir sensu stricto) by microsatellite markers and mitochondrial COI gene sequence. Gene 2019, 20, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.R.; Zhang, X.Y.; Yang, X.X.; Liao, W.J. Development of polymorphic microsatellite markers for Fagus pashanica (Fagaceae) using next-generation sequencing. Genes Genet. Syst. 2023, 21, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhu, W.; Wu, Y.; Wei, S.; Shu, G.; Tian, Y.; Du, X.; Tang, J.; Feng, Y.; Wu, G.; et al. Whole-genome sequencing revealed genetic diversity, structure and patterns of selection in Guizhou indigenous chickens. BMC Genom. 2023, 26, 24570. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Schaffner, S.F.; Fry, B.; Lohmueller, J.; Varilly, P.; Shamovsky, O.; Palma, A.; Mikkelsen, T.S.; Altshuler, D.; Lander, E.S. Positive natural selection in the human lineage. Science 2006, 16, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Li, H.F.; Zhu, W.Q.; Chen, K.W.; Xu, W.J.; Song, W. Two maternal origins of Chinese domestic goose. Poult. Sci. 2011, 90, 2705–2710. [Google Scholar] [CrossRef] [PubMed]
- Honka, J.; Heino, M.T.; Kvist, L.; Askeyev, I.V.; Shaymuratova, D.N.; Askeyev, O.V.; Askeyev, A.O.; Heikkinen, M.E.; Searle, J.B.; Aspi, J. Over a Thousand Years of Evolutionary History of Domestic Geese from Russian Archaeological Sites, Analysed Using Ancient DNA. Genes 2018, 20, 361–367. [Google Scholar] [CrossRef]
- Ran, B.; Zhu, W.; Zhao, X.; Li, L.; Yi, Z.; Li, M.; Wang, T.; Li, D. Studying Genetic Diversity and Relationships between Mountainous Meihua Chickens Using Mitochondrial DNA Control Region. Genes 2023, 28, 145998. [Google Scholar] [CrossRef] [PubMed]
- Du-plessis, S.J.; Blaxter, M.; Koepfli, K.P.; Chadwick, E.A.; Hailer, F. Genomics Reveals Complex Population History and Unexpected Diversity of Eurasian Otters (Lutra lutra) in Britain Relative to Genetic Methods. Mol. Biol. Evol. 2023, 40, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, T. Irregularities in genetic variation and mutation rates with environmental stresses. Environ. Microbiol. 2019, 21, 3979–3988. [Google Scholar] [CrossRef]
- Ishibashi, Y. Preservation of genetic diversity in a highly fragmented population of the gray-sided vole Myodes rufocanus in an intensive farming region. Ecol. Evol. 2023, 19, 310472. [Google Scholar] [CrossRef]
- Honka, J.; Baini, S.; Searle, J.B.; Kvist, L.; Aspi, J. Genetic assessment reveals inbreeding, possible hybridization, and low levels of genetic structure in a declining goose population. Ecol. Evol. 2022, 12, e8547. [Google Scholar] [CrossRef]
- Ma, Z.; Feng, M.; Tang, Y. Genetic diversity of serrated rice shrimp population in Henan Province based on mitochondrial COI gene. Sichuan Zool. 2023, 42, 481–489. [Google Scholar]
- Boman, J.; Mugal, C.F.; Backström, N. The Effects of GC-Biased Gene Conversion on Patterns of Genetic Diversity among and across Butterfly Genomes. Genome Biol. Evol. 2021, 7, 1059–1064. [Google Scholar] [CrossRef]
- Lamichhaney, S.; Han, F.; Webster, M.T.; Grant, B.R.; Grant, P.R.; Andersson, L. Female-biased gene flow between two species of Darwin’s finches. Nat. Ecol. Evol. 2020, 4, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.; Koc, G.; Eyre-Walker, Y.; Eyre-Walker, A. What Determines Levels of Mitochondrial Genetic Diversity in Birds? Genome Biol. Evol. 2023, 15, 521064. [Google Scholar] [CrossRef]
- Shi, X.W.; Wang, J.W.; Zeng, F.T.; Qiu, X.P. Mitochondrial DNA cleavage patterns distinguish independent origin of Chinese domestic geese and Western domestic geese. Biochem. Genet. 2006, 44, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Dong, S.; Zhao, Y.; Yang, L.; Qi, X.; Ren, Z.; Dong, S.; Cheng, J. Genetic diversity, population genetic structure and gene flow in the rare and endemic endangered wild plant Cypripedium macranthos revealed by genotyping-by-sequencing. BMC Plant Biol. 2023, 15, 231254. [Google Scholar]
- Robinson, J.; Kyriazis, C.C.; Yuan, S.C.; Lohmueller, K.E. Deleterious Variation in Natural Breeds and Implications for Conservation Genetics. Annu. Rev. Anim. Biosci. 2023, 15, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, M.E.; Ruokonen, M.; White, T.A.; Alexander, M.M.; Gündüz, İ.; Dobney, K.M.; Aspi, J.; Searle, J.B.; Pyhäjärvi, T. Long-Term Reciprocal Gene Flow in Wild and Domestic Geese Reveals Complex Domestication History. G3 2020, 10, 3061–3070. [Google Scholar] [CrossRef]
- Wójcik, E.; Smalec, E. Constitutive heterochromatin in chromosomes of duck hybrids and goose hybrids. Poult. Sci. 2017, 96, 18–26. [Google Scholar] [CrossRef]
- McKinney, M.L. Mass Extinctions. Processes and Evidence. Stephen K. Donovan, Ed. Columbia University Press, New York. Science 1990, 26, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qi, S.; Liu, L.; Bao, Q.; Wu, T.; Liu, W.; Zhang, Y.; Zhao, W.; Xu, Q.; Chen, G. Genetic Diversity Analysis and Breeding of Geese Based on the Mitochondrial ND6 Gene. Genes 2023, 10, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Song, J.; Wang, X. Genetic diversity of fine scaled fish breeds in the Yalu River Basin based on mitochondrial CYTB gene and D-loop region. Northwest Agric. J. 2023, 32, 1675–1685. [Google Scholar]
- Souza, T.O.; Luna, L.W.; Araripe, J.; Silva, W.; Rego, P. Peripheral isolation and demographic stability are reflected in the genetic diversity of the breeds of the Helmeted Manakin: A bird endemic to the gallery forests. An. Acad. Bras. Cienc. 2022, 16, 20201206. [Google Scholar] [CrossRef]
- Du, W. A Study on the Diversity and Taxonomic Status of Genetic Resources of Local Goose Species in China. Nanjing Agric. Univ. 2012, 15, 568–599. [Google Scholar]
- Ham-Dueñas, J.G.; Canales-Del-Castillo, R.; Voelker, G.; Ruvalcaba-Ortega, I.; Aguirre-Calderón, C.E.; González-Rojas, J.I. Adaptive genetic diversity and evidence of population genetic structure in the endemic endangered Sierra Madre Sparrow (Xenospiza baileyi). PLoS ONE 2020, 30, e0232282. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Birthplace | Appearance Characteristics | Number/w | Protection Level |
|---|---|---|---|---|
| LX | Zhuzhou, Hunan | Compact body shape and pure white feathers | 0.04 | Endemic endangered |
| YE | Lu’an, Anhui | Flat sarcoma with black and gray feathers | 0.09 | Endemic endangered |
| YJ | Yangjiang, Guangdong | Large body size and pure white feathers | 0.09 | Endemic endangered |
| WZ | Qingyuan, Guangdong | Black mane with back feathers and black-gray feathers | 0.20 | Endemic endangered |
| XP | Xupu, Hunan | Head sarcoma and black-gray feathers | 0.60 | Dangerous |
| BZ | Jinxiang, Shandong | Wide and short body with pure white feathers | 1.00 | Dangerous |
| Breed | NPIS | PIS | MS | GC | Hd | Pi | K |
|---|---|---|---|---|---|---|---|
| BZ | 1 | 10 | 646 | 48.87% | 0.952 ± 0.0230 | 0.00538 ± 0.00054 | 3.538 ± 0.081 |
| LX | 1 | 17 | 642 | 48.97% | 0.984 ± 0.0120 | 0.00910 ± 0.00162 | 6.005 ± 0.138 |
| XP | 0 | 18 | 621 | 48.94% | 0.985 ± 0.0120 | 0.00806 ± 0.00106 | 5.153 ± 0.123 |
| YJ | 4 | 18 | 629 | 48.99% | 0.943 ± 0.0200 | 0.01300 ± 0.00060 | 8.463 ± 0.202 |
| YE | 3 | 20 | 633 | 49.27% | 0.993 ± 0.0110 | 0.01137 ± 0.00112 | 7.457 ± 0.171 |
| WZ | 8 | 17 | 614 | 48.78% | 0.956 ± 0.0240 | 0.01307 ± 0.00199 | 8.350 ± 0.199 |
| Breed | h | Unique Haplotypes | Proportion |
|---|---|---|---|
| BZ | 19 | 7 ♀: Hap_74, Hap_75, Hap_76, Hap_77, Hap_78, Hap_79, Hap_80 | 42.11% |
| 1 ♂: Hap_81 | |||
| LX | 23 | 7 ♀: Hap_63, Hap_64, Hap_65, Hap_66, Hap_67, Hap_68, Hap_69 | 39.13% |
| 2 ♂: Hap_70, Hap_71 | |||
| XP | 23 | 6 ♀: Hap_37, Hap_38, Hap_39, Hap_40, Hap_41, Hap_42, | 47.83% |
| 5 ♂: Hap_43, Hap_44, Hap_45, Hap_47, Hap_49 | |||
| YJ | 15 | 4 ♀: Hap_2, Hap_5, Hap_7, Hap_8 | 40.00% |
| 2 ♂: Hap_12, Hap_15 | |||
| YE | 27 | 7 ♀: Hap_17, Hap_18, Hap_20, Hap_21, Hap_22, Hap_24, Hap_27 | 37.04% |
| 3 ♂: Hap_31, Hap_32, Hap_36 | |||
| WZ | 20 | 8 ♀: Hap_50, Hap_51, Hap_52, Hap_53, Hap_54, Hap_55, Hap_56, Hap_57, | 60.00% |
| 4 ♂: Hap_58, Hap_59, Hap_61, Hap_62 |
| Breed | BZ | LX | XP | YJ | YE | WZ |
|---|---|---|---|---|---|---|
| BZ | 0.090 | 0.000 | 1.400 | 6.220 | 1.390 | |
| LX | 0.852 | −27.140 | 3.840 | −22.730 | 15.860 | |
| XP | 0.991 | 0.019 | 2.770 | −62.450 | 7.330 | |
| YJ | 0.263 | 0.115 | 0.153 | 3.050 | 70.330 | |
| YE | 0.074 | 0.023 | 0.008 | 0.076 | 31.880 | |
| WZ | 0.153 | 0.031 | 0.064 | 0.004 | 0.015 |
| Breed | DF | SS | MS | EVV | PV | Fst | p Value |
|---|---|---|---|---|---|---|---|
| AP | 5.0 | 51.483 | 67.354 | 0.24179 | 0.071 | 0.07099 | 0.001 |
| WP | 171.0 | 541.099 | 32.646 | 3.16432 | 0.929 | - | - |
| Total | 176.0 | 592.582 | 100.000 | 3.40611 | 1.000 | 0.07099 | 0.001 |
| Item | BZ | LX | XP | YJ | YE | WZ |
|---|---|---|---|---|---|---|
| Fu’s Fs | −11.559 | −13.025 | −15.668 | −1.020 | −19.317 | −5.615 |
| p-value | p > 0.10 | p > 0.10 | p < 0.02 | p > 0.10 | p > 0.10 | p > 0.10 |
| Tajima’ D | 0.88243 | 1.10756 | 0.43066 | 1.80847 | 1.00525 | 0.93890 |
| p-value | p > 0.10 | p > 0.10 | p > 0.10 | 0.1 > p > 0.05 | p > 0.10 | p > 0.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, S.; Fan, S.; Li, H.; He, Y.; Zhang, Y.; Zhao, W.; Xu, Q.; Chen, G. Analysis of Genetic Diversity and Population Structure of Endemic Endangered Goose (Anser cygnoides) Breeds Based on Mitochondrial CYTB. Animals 2024, 14, 1480. https://doi.org/10.3390/ani14101480
Qi S, Fan S, Li H, He Y, Zhang Y, Zhao W, Xu Q, Chen G. Analysis of Genetic Diversity and Population Structure of Endemic Endangered Goose (Anser cygnoides) Breeds Based on Mitochondrial CYTB. Animals. 2024; 14(10):1480. https://doi.org/10.3390/ani14101480
Chicago/Turabian StyleQi, Shangzong, Suyu Fan, Haoyu Li, Yufan He, Yang Zhang, Wenming Zhao, Qi Xu, and Guohong Chen. 2024. "Analysis of Genetic Diversity and Population Structure of Endemic Endangered Goose (Anser cygnoides) Breeds Based on Mitochondrial CYTB" Animals 14, no. 10: 1480. https://doi.org/10.3390/ani14101480
APA StyleQi, S., Fan, S., Li, H., He, Y., Zhang, Y., Zhao, W., Xu, Q., & Chen, G. (2024). Analysis of Genetic Diversity and Population Structure of Endemic Endangered Goose (Anser cygnoides) Breeds Based on Mitochondrial CYTB. Animals, 14(10), 1480. https://doi.org/10.3390/ani14101480