
Composition of the Midgut Microbiota Structure of Haemaphysalis longicornis Tick Parasitizing Tiger and Deer


Abstract
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
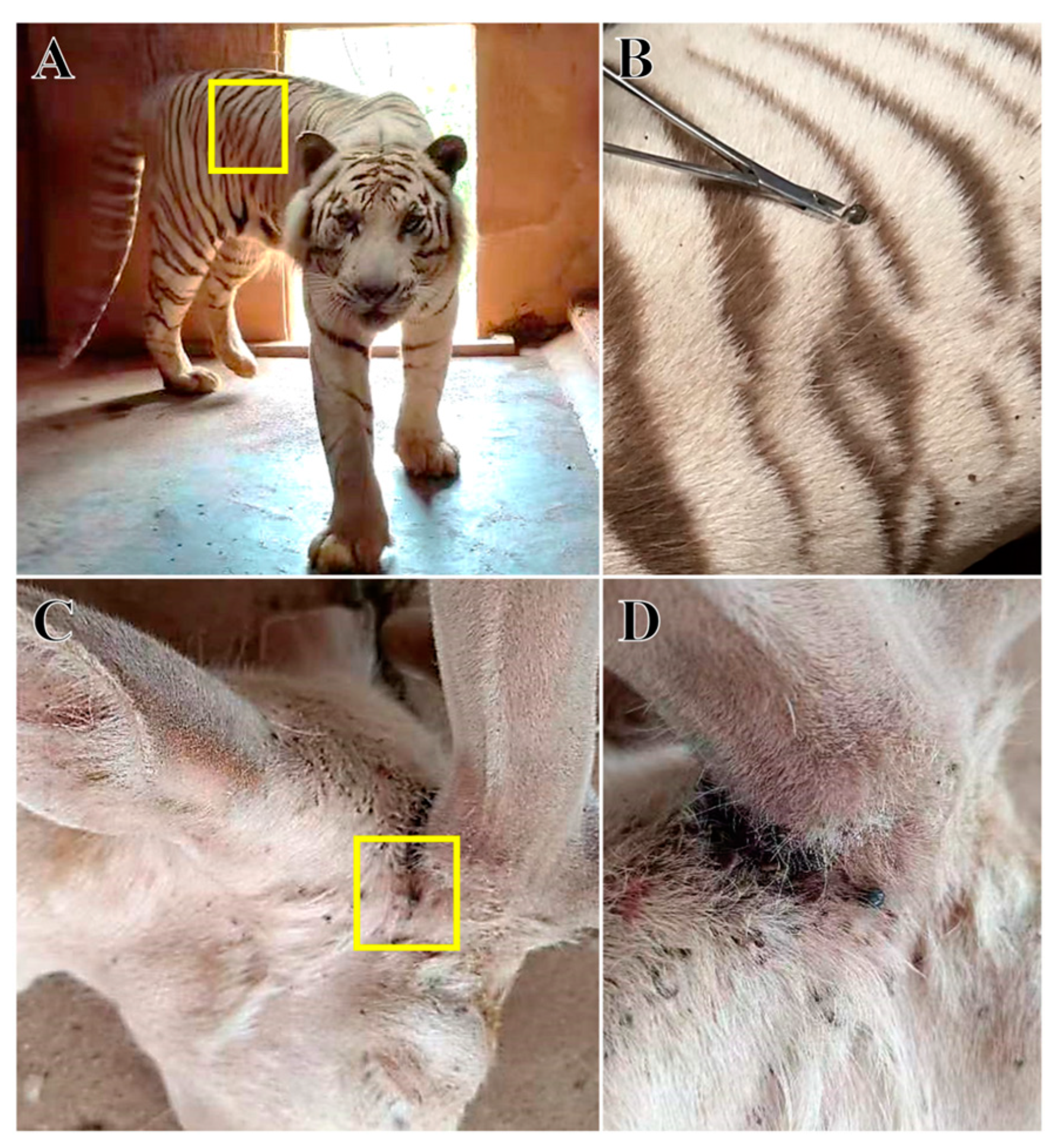
2.1. Collection and Identification of Tick Samples
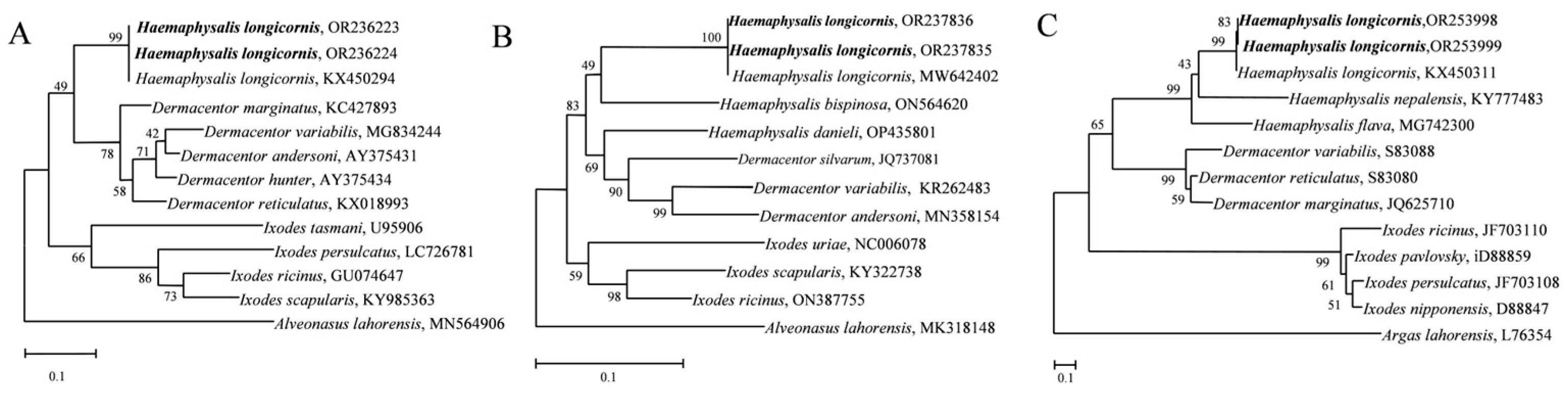
2.2. Identification of Tick Species
2.3. DNA Extraction of the Midgut Microbiota and Blood Extraction
2.4. 16S rRNA Gene Amplification and High-Throughput Sequencing
2.5. Data Analysis
2.5.1. Sequencing Data Processing
2.5.2. Amplicon Sequence Variant Noise Reduction and Species Annotation
2.5.3. Alpha Diversity Analysis
2.5.4. Beta Diversity Analysis
2.5.5. Identification of Morganella in Ticks
3. Results
3.1. Identification of Ticks
3.2. Data Processing and ASVs Statistical Results
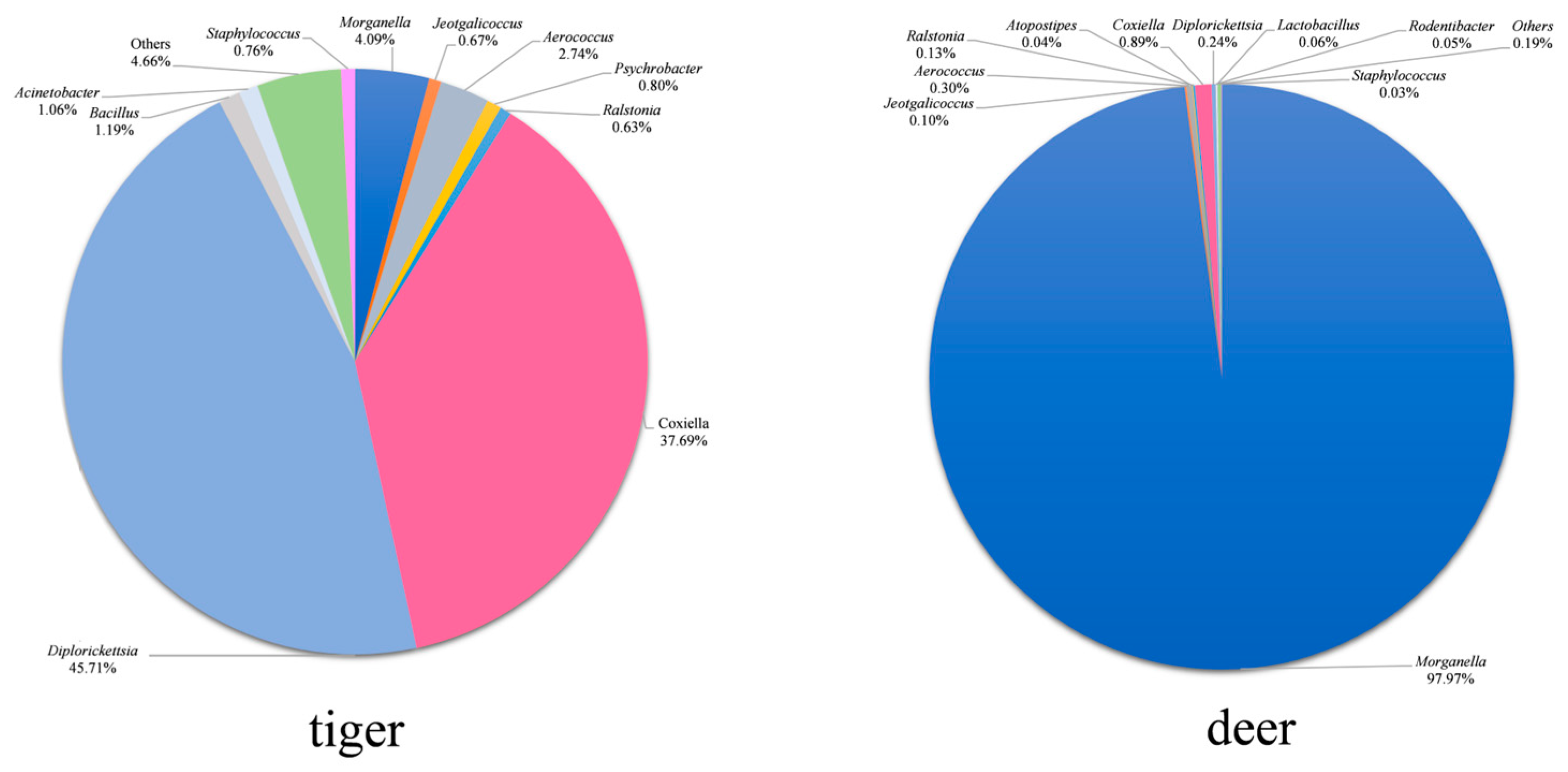
3.3. Analysis of Microbial Compositions
3.4. Difference Analysis of Midgut Microbiota Structure
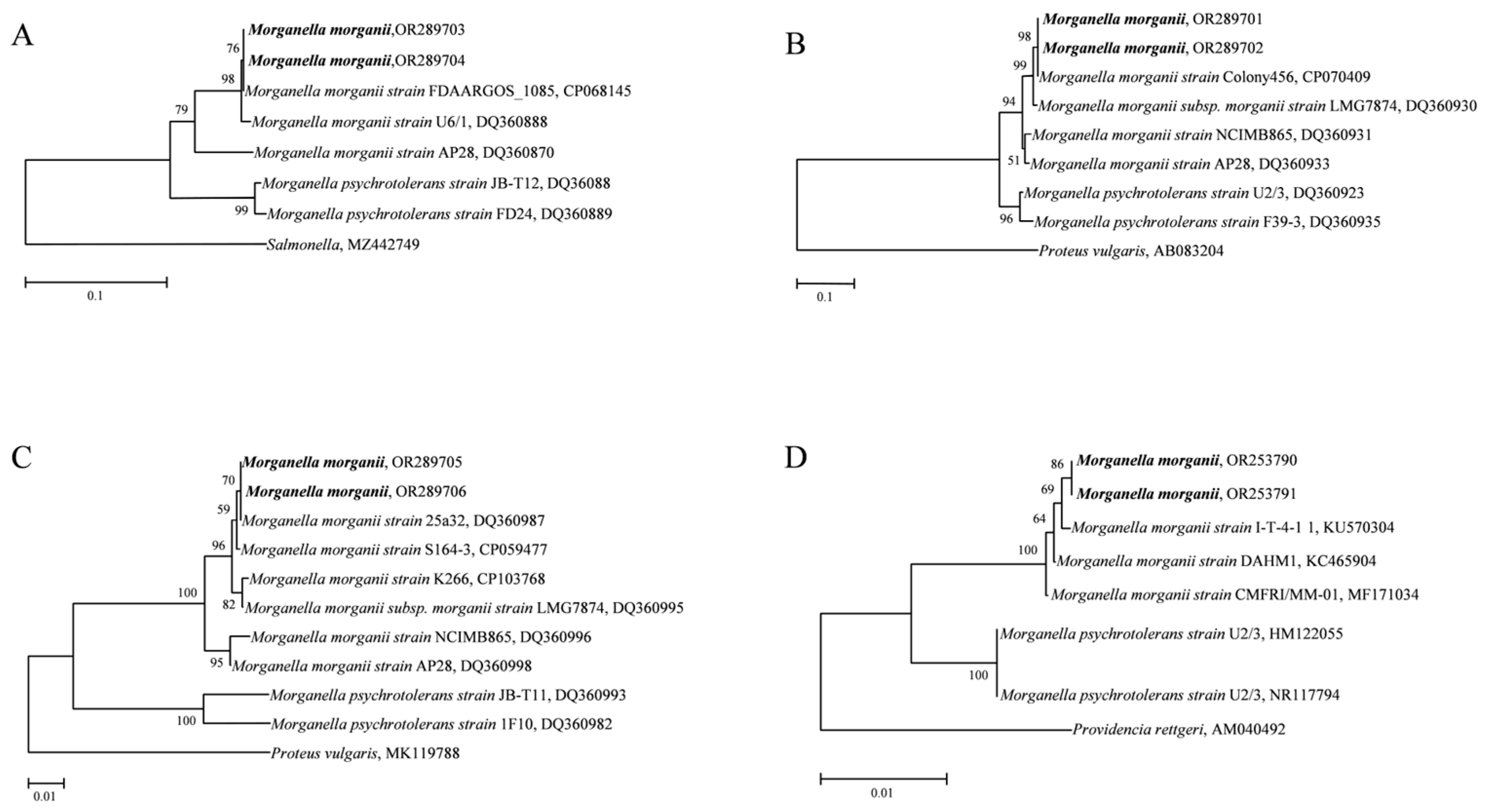
3.5. Identification of Morganella
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pritt, B.S. Haemaphysalis longicornis is in the United States and biting humans: Where do we go from here? Clin. Infect. Dis. 2020, 70, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Rainey, T.; Occi, J.L.; Robbins, R.G.; Egizi, A. Discovery of Haemaphysalis longicornis (Ixodida: Ixodidae) parasitizing a sheep in New Jersey, United States. J. Med. Entomol. 2018, 55, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Heath, A. Biology, ecology and distribution of the tick, Haemaphysalis longicornis Neumann (Acari: Ixodidae) in New Zealand. N. Z. Vet. J. 2016, 64, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Haji, I.; Simuunza, M.; Jiang, N.; Chen, Q. Tick populations and molecular detection of selected tick-borne pathogens in questing ticks from northern and central Tanzania. Exp. Appl. Acarol. 2023, 90, 389–407. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, A.M.; Brinkerhoff, R.J.; Bernal, J.; Bermúdez, S.E. Ticks and tick-borne pathogens of dogs along an elevational and land-use gradient in Chiriquí province, Panamá. Exp. Appl. Acarol. 2017, 71, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Talactac, M.R.; Yoshii, K.; Hernandez, E.P.; Kusakisako, K.; Galay, R.L.; Fujisaki, K.; Mochizuki, M.; Tanaka, T. Synchronous Langat virus infection of Haemaphysalis longicornis using anal pore microinjection. Viruses 2017, 9, 189. [Google Scholar] [CrossRef]
- Zhuang, L.; Sun, Y.; Cui, X.; Tang, F.; Hu, J.; Wang, L.; Cui, N.; Yang, Z.; Huang, D.; Zhang, X.; et al. Transmission of severe fever with thrombocytopenia syndrome virus by Haemaphysalis longicornis ticks. China Emerg. Infect. Dis. 2018, 24, 868–871. (In Chinese) [Google Scholar] [CrossRef]
- Qin, X.; Han, F.; Luo, L.; Zhao, F.; Han, H.; Zhang, Z.; Liu, J.; Xue, Z.; Liu, M.; Ma, D.; et al. Anaplasma species detected in Haemaphysalis longicornis tick from China. Ticks Tick Borne Dis. 2018, 9, 840–843. [Google Scholar] [CrossRef]
- Stanley, H.M.; Ford, S.L.; Snellgrove, A.N.; Hartzer, K.; Smith, E.B.; Krapiunaya, I.; Levin, M.L. The ability of the invasive Asian longhorned tick Haemaphysalis longicornis (Acari: Ixodidae) to acquire and transmit Rickettsia rickettsii (Rickettsiales: Rickettsiaceae), the agent of Rocky Mountain spotted fever, under laboratory conditions. J. Med. Entomol. 2020, 57, 1635–1639. [Google Scholar] [CrossRef]
- Cirimotich, C.M.; Ramirez, J.L.; Dimopoulos, G. Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe 2011, 10, 307–310. [Google Scholar] [CrossRef]
- Weiss, B.; Aksoy, S. Microbiome influences on insect host vector competence. Trends Parasitol. 2011, 27, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.Q.; Chen, J.W.; Xia, M.Y. Growth kinetics of systemic Lyme disease spirochetes in blood-feeding Ixodes ricinus females. Chin. J. Zoonoses 1998, 14, 16–20. (In Chinese) [Google Scholar]
- Dillon, R.J.; Dillon, V.M. The gut bacteria of insects: Nonpathogenic interactions. Annu. Rev. Entomol. 2004, 49, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Huang, Z.; Yu, G.; Zhang, Z. Characterization of the bacterial community in Haemaphysalis longicornis (Acari: Ixodidae) throughout developmental stages. Exp. Appl. Acarol. 2019, 77, 173–186. [Google Scholar]
- Li, S.; Zhang, X.; Zhou, X.; Chen, K.; Masoudi, A.; Liu, J.; Zhang, Y. Bacterial microbiota analysis demonstrates that ticks can acquire bacteria from habitat and host blood meal. Exp. Appl. Acarol. 2022, 87, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Lv, Q.; Chang, Q.; Ju, H.; Wu, T.; Liu, S.; Li, X.; Yan, Y.; Gao, J.; Wang, C. Microbiota Community Structure and Interaction Networks within Dermacentor silvarum, Ixodes persulcatus, and Haemaphysalis concinna. Animals 2022, 12, 3237. [Google Scholar] [CrossRef] [PubMed]
- Teng, G.F.; Jiang, Z.J. Acari: Ixodidae. Economicinsect Fauna of China; Science Press: Beijing, China, 1991. (In Chinese) [Google Scholar]
- DUAN, D.; LIU, G.; CHENG, T. Morphological and molecular markers characteristics of Argas persicus. Chin. Vet. Sci. 2019, 49, 990–997. (In Chinese) [Google Scholar]
- Zhou, H.; Cheng, T.; Duan, D. Comparison of morphological structure and molecular markers between Dermacentor nuttal and Dermacentor silvarum. Chin. Vet. Sci. 2019, 49, 317–323. (In Chinese) [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory-Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R. Reproducible, interactive, scalable, and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Emborg, J.; Dalgaard, P.; Ahrens, P. Morganella psychrotolerans sp. nov., a histamine-producing bacterium isolated from various seafoods. Int. J. Syst. Evol. Microbiol. 2006, 56, 2473–2479. [Google Scholar] [CrossRef]
- Guizzo, M.G.; Dolezelikova, K.; Neupane, S.; Frantova, H.; Hrbatova, A.; Pafco, B.; Fiorotti, J.; Kopacek, P.; Zurek, L. Characterization and manipulation of the bacterial community in the midgut of Ixodes ricinus. Parasites Vectors 2022, 15, 248. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.I.; Binetruy, F.; Hernández-Jarguín, A.M.; Duron, O. The tick microbiome: Why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front. Cell. Infect. Microbiol. 2017, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Ohtaka, C.; Ishikawa, H. Effects of heat treatment on the symbiotic system of an aphid mycetocyte. Symbiosis 1991, 11, 19–30. [Google Scholar]
- Smith, T.A.; Driscoll, T.; Gillespie, J.J.; Raghavan, R. A Coxiella-like endosymbiont is a potential vitamin source for the Lone Star tick. Genome Biol. Evol. 2015, 7, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Greay, T.L.; Evasco, K.L.; Evans, M.L.; Oskam, C.L.; Magni, P.A.; Ryan, U.M.; Irwin, P.J. Illuminating the bacterial microbiome of Australian ticks with 16S and Rickettsia-specific next-generation sequencing. Curr. Res. Parasitol. Vector Borne Dis. 2021, 1, 100037. [Google Scholar] [CrossRef]
- Grostieta, E.; Zazueta-Islas, H.M.; Cruz-Valdez, T.; Ballados-González, G.G.; Álvarez-Castillo, L.; García-Esparza, S.M.; Cruz-Romero, A.; Romero-Salas, D.; Aguilar-Domínguez, M.; Becker, I.; et al. Molecular detection of Coxiella-like endosymbionts and absence of Coxiella burnetii in Amblyomma mixtum from Veracruz, Mexico. Exp. Appl. Acarol. 2022, 88, 113–125. [Google Scholar] [CrossRef]
- Abdel-Moein, K.A.; Hamza, D.A. The burden of Coxiella burnetii among aborted dairy animals in Egypt and its public health implications. Acta Trop. 2017, 166, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.W.; Derrick, E.H. Studies in the epidemiology of Q fever: 1. The isolation of six strains of Rickettsia burneti from the tick Haemaphysalis humerosa. Aust. J. Exp. Biol. Med. Sci. 1940, 18, 1–8. [Google Scholar] [CrossRef]
- Borawski, K.; Dunaj, J.; Czupryna, P.; Pancewicz, S.; Świerzbińska, R.; Żebrowska, A.; Moniuszko-Malinowska, A. Assess-ment of Coxiella burnetii presence after tick bite in north-eastern Poland. Infection 2020, 48, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Klyachko, O.; Stein, B.D.; Grindle, N.; Clay, K.; Fuqua, C. Localization and visualization of a Coxiella-type symbiont within the lone star tick, Amblyomma americanum. Appl. Environ. Microbiol. 2007, 73, 6584–6594. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.P.; Marcili, A.; Leite, R.C.; Nieri-Bastos, F.A.; Domingues, L.N.; Martins, J.R.; Labruna, M.B. Coxiella symbiont in the tick Ornithodoros rostratus (Acari: Argasidae). Ticks Tick Borne Dis. 2012, 3, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Lalzar, I.; Friedmann, Y.; Gottlieb, Y. Tissue tropism and vertical transmission of Coxiella in Rhipicephalus sanguineus and Rhipicephalus turanicus ticks. Environ. Microbiol. 2014, 16, 3657–3668. [Google Scholar] [CrossRef]
- Duron, O.; Jourdain, E.; McCoy, K.D. Diversity and global distribution of the Coxiella intracellular bacterium in seabird ticks. Ticks Tick Borne Dis. 2014, 5, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, S.; Chen, K.; Yang, C.; Zhou, X.; Liu, J.; Zhang, Y. Growth dynamics and tissue localization of a Coxiella-like endosymbiont in the tick Hemaphysalis longicornis. Ticks Tick Borne Dis. 2022, 13, 102005. [Google Scholar] [CrossRef] [PubMed]
- Guizzo, M.G.; Parizi, L.F.; Nunes, R.D.; Schama, R.; Albano, R.M.; Tirloni, L.; Oldiges, D.P.; Vieira, R.P.; Oliveira, W.H.C.; Leite, M.S.; et al. A Coxiella mutualist symbiont is essential to the development of Rhipicephalus microplus. Sci. Rep. 2017, 14, 17554. [Google Scholar] [CrossRef]
- Duron, O.; Gottlieb, Y. Convergence of Nutritional Symbioses in Obligate Blood Feeders. Trends Parasitol. 2020, 36, 816–825. [Google Scholar] [CrossRef]
- Buysse, M.; Duron, O. Evidence that microbes identified as tick-borne pathogens are nutritional endosymbionts. Cell 2021, 29, 2259–2260. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X. Study on Molecular Biological Strain Identification of Acinetobacter and Its Clinical Application. Master’s Thesis, University of South China, Hengyang, China, 2018. (In Chinese). [Google Scholar]
- Murrell, A.; Dobson, S.J.; Yang, X.; Lacey, E.; Barker, S.C. A survey of bacterial diversity in ticks, lice and fleas from Australia. Parasitol. Res. 2003, 89, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.A.; Gall, C.A.; Mason, K.L.; Scoles, G.A.; Brayton, K.A. The characterization and manipulation of the bacterial microbiome of the Rocky Mountain wood tick, Dermacentor andersoni. Parasites Vectors 2015, 8, 632. [Google Scholar] [CrossRef] [PubMed]
- Piette, A.; Verschraegen, G. Role of coagulase-negative Staphylococci in human disease. Vet. Microbiol. 2009, 134, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, Y.; Luo, X. Analysis of bacterial diversity of ticks in Yuyao City, Zhejiang Province based on high-throughput sequencing technology. Dis. Surveill. 2019, 35, 642–645. (In Chinese) [Google Scholar]
- Budachetri, K.; Williams, J.; Mukherjee, N.; Sellers, M.; Moore, F.; Karim, S. The microbiome of neotropical ticks parasitizing on passerine migratory birds. Ticks Tick Borne Dis. 2017, 8, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Menchaca, A.C.; Visi, D.K.; Strey, O.F.; Teel, P.D.; Kalinowski, K.; Allen, M.S.; Williamson, P.C. Preliminary assessment of microbiome changes following blood-feeding and survivorship in the Amblyomma americanum nymph-to-adult transition using semiconductor Sequencing. PLoS ONE 2013, 8, e67129. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ai, L.; Zhu, C.; Lu, Y.; Lv, R.; Mao, Y.; Lu, N.; Tan, W. Co-existence of Multiple Anaplasma Species and Variants in Ticks Feeding on Hedgehogs or Cattle Poses Potential Threats of Anaplasmosis to Humans and Livestock in Eastern China. Front. Microbiol. 2022, 13, 913650. [Google Scholar] [CrossRef] [PubMed]
- Garcês, A.; Poeta, P.; Soeiro, V.; Lóio, S.; Cardoso-Gomes, A.; Torres, C.; Pires, I. Pyometra Caused by Staphylococcus lentus in a Wild European Hedgehog (Erinaceus europaeus). J. Wildl. Dis. 2019, 55, 724–727. [Google Scholar] [CrossRef]
- Guerra, M.; Marado, D. Fulminans Purpura due to Morganella morganii. Eur. J. Case Rep. Intern. Med. 2022, 9, 003670. [Google Scholar] [CrossRef]
- Podeur, G.; Dalgaard, P.; Leroi, F.; Prévost, H.; Emborg, J.; Martinussen, J.; Hansen, L.H.; Pilet, M.F. Development of a real-time PCR method coupled with a selective pre-enrichment step for quantification of Morganella morganii and Morganella psychrotolerans in fish products. Int. J. Food Microbiol. 2015, 203, 55–62. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, C.M.; Brenner, F.W.; Miller, J.M. Classification, identification, and clinical significance of Proteus, Providencia, and Morganella. Clin. Microbiol. Rev. 2000, 13, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Briceño, C.; González-Acuña, D.; Jiménez, J.E.; Bornscheuer, M.L.; Funk, S.M.; Knapp, L.A. Ear Mites, Otodectes Cynotis, on Wild Foxes (Pseudalopex spp.) in Chile. J. Wildl. Dis. 2020, 56, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Klubal, R.; Kopecky, J.; Nesvorna, M.; Sparagano, O.A.; Thomayerova, J.; Hubert, J. Prevalence of pathogenic bacteria in Ixodes ricinus ticks in Central Bohemia. Exp. Appl. Acarol. 2016, 68, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.L.; Cheng, T.Y.; Yang, H.; Yan, F. Identification of intestinal bacterial flora in Rhipicephalus microplus ticks by conventional methods and PCR-DGGE analysis. Exp. Appl. Acarol. 2015, 66, 257–268. [Google Scholar] [CrossRef]
- Segura, J.A.; Isaza, J.P.; Botero, L.E.; Alzate, J.F.; Gutiérrez, L.A. Assessment of bacterial diversity of Rhipicephalus microplus ticks from two livestock agroecosystems in Antioquia, Colombia. PLoS ONE 2020, 15, e0234005. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Names | Primer Sequences (5′→3′) |
|---|---|
| 16S rDNA-forward 1 | 5′-CTGCTCAATGATTTTTTAAATTGCTGTGG-3′ |
| 16S rDNA-reverse 1 | 5′-CCGGTCTGAACTCAGATCAAGT-3′ |
| COX1-forward | 5′-GGAACAATATATTTAATTTTTGG-3′ |
| COX1-reverse | 5′ATCTATCCCTACTGTAAATATATG-3′ |
| ITS-forward | 5′-CGAGACTTGGTGTGAATTGCA-3′ |
| ITS-reverse | 5′-TCCCATACACCACATTTCCCG-3′ |
| 16S rDNA-forward 2 | 5′-AGAGTTTGATCCTGGCTCAG-3′ |
| 16S rDNA-reverse 2 | 5′-GGTTACCTTGTTACGACTT-3′ |
| dnaN-foward | 5′-ATGAAATTTACCGTTGAACGTGA-3′ |
| dnaN-reverse | 5′-CCATCCACCAGCTTCGAGGT-3′ |
| tuf-foward | 5′-TTGTTGCTGCAACTGATGG-3′ |
| tuf-reverse | 5′-CACCTTCATCTTTGCTCAG-3′ |
| hdc-foward | 5′-TCHATYARYAACTGYGGTGACTGGRG-3′ |
| hdc-reverse | 5′-CCCACAKCATBARWGGDGTRTGRC-3′ |
| Group | Chao1 | Good’s Coverage | Shannon | Simpson |
|---|---|---|---|---|
| Deer host | 31.000 | 1.000 | 1.526 | 0.573 |
| Tiger host | 138.000 | 1.000 | 2.319 | 0.647 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.-L.; Qiu, Q.-G.; Cheng, T.-Y.; Liu, G.-H.; Liu, L.; Duan, D.-Y. Composition of the Midgut Microbiota Structure of Haemaphysalis longicornis Tick Parasitizing Tiger and Deer. Animals 2024, 14, 1557. https://doi.org/10.3390/ani14111557
Liu Z-L, Qiu Q-G, Cheng T-Y, Liu G-H, Liu L, Duan D-Y. Composition of the Midgut Microbiota Structure of Haemaphysalis longicornis Tick Parasitizing Tiger and Deer. Animals. 2024; 14(11):1557. https://doi.org/10.3390/ani14111557
Chicago/Turabian StyleLiu, Zi-Ling, Qi-Guan Qiu, Tian-Yin Cheng, Guo-Hua Liu, Lei Liu, and De-Yong Duan. 2024. "Composition of the Midgut Microbiota Structure of Haemaphysalis longicornis Tick Parasitizing Tiger and Deer" Animals 14, no. 11: 1557. https://doi.org/10.3390/ani14111557
APA StyleLiu, Z.-L., Qiu, Q.-G., Cheng, T.-Y., Liu, G.-H., Liu, L., & Duan, D.-Y. (2024). Composition of the Midgut Microbiota Structure of Haemaphysalis longicornis Tick Parasitizing Tiger and Deer. Animals, 14(11), 1557. https://doi.org/10.3390/ani14111557