

Diet Drives Gut Bacterial Diversity of Wild and Semi-Captive Common Cranes (Grus grus)

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fecal Sample Collection and DNA Extraction
2.2. DNA Extraction and Sequencing
2.3. Bioinformatics Analysis
2.4. Diet and Microhistological Analysis
3. Results
3.1. Microbial Community Diversity and Alpha Diversity
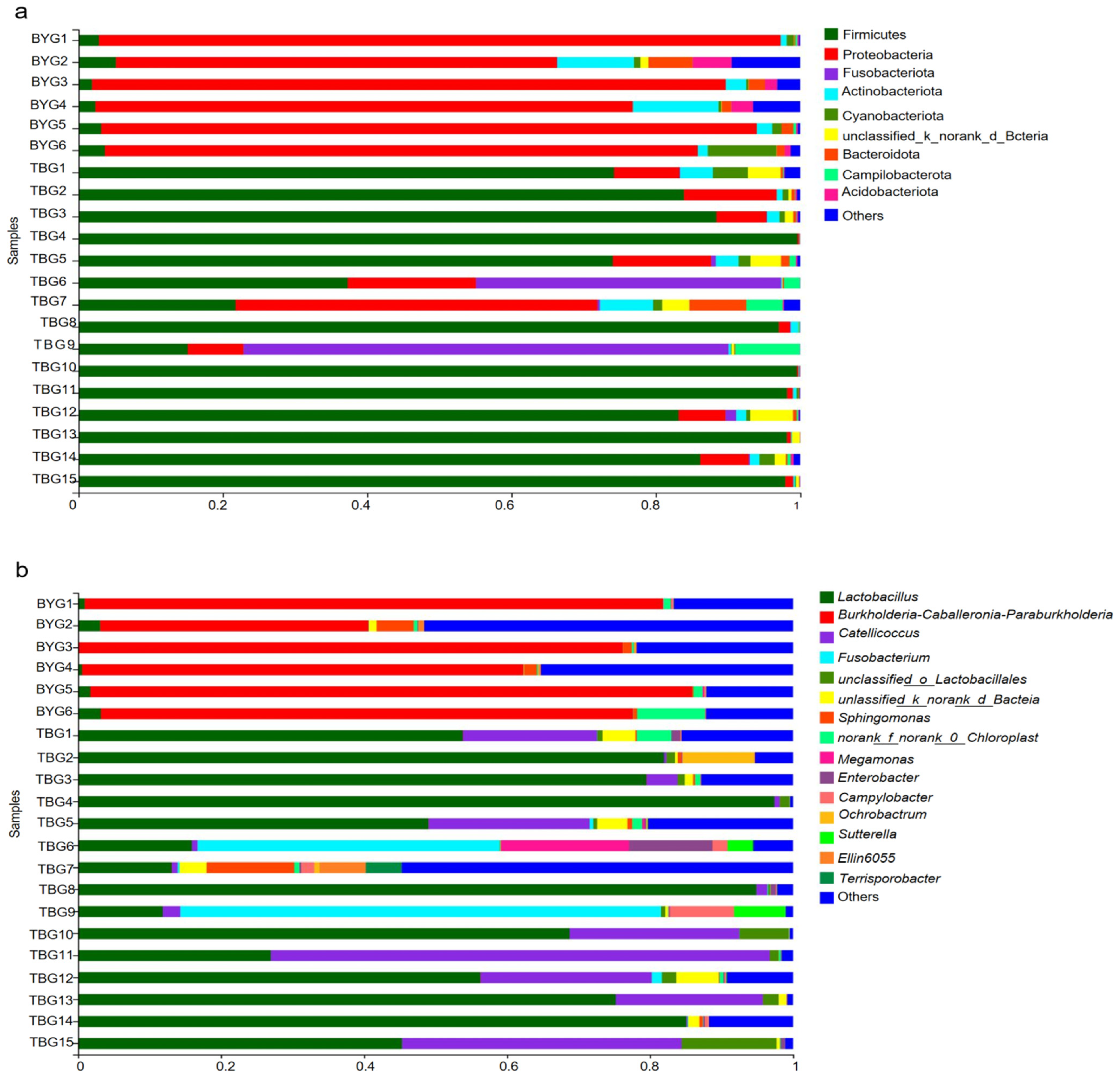
3.2. Taxonomic Comparisons of Gut Microbiota between Two Groups
3.3. Beta Diversity of G. grus from Different Groups
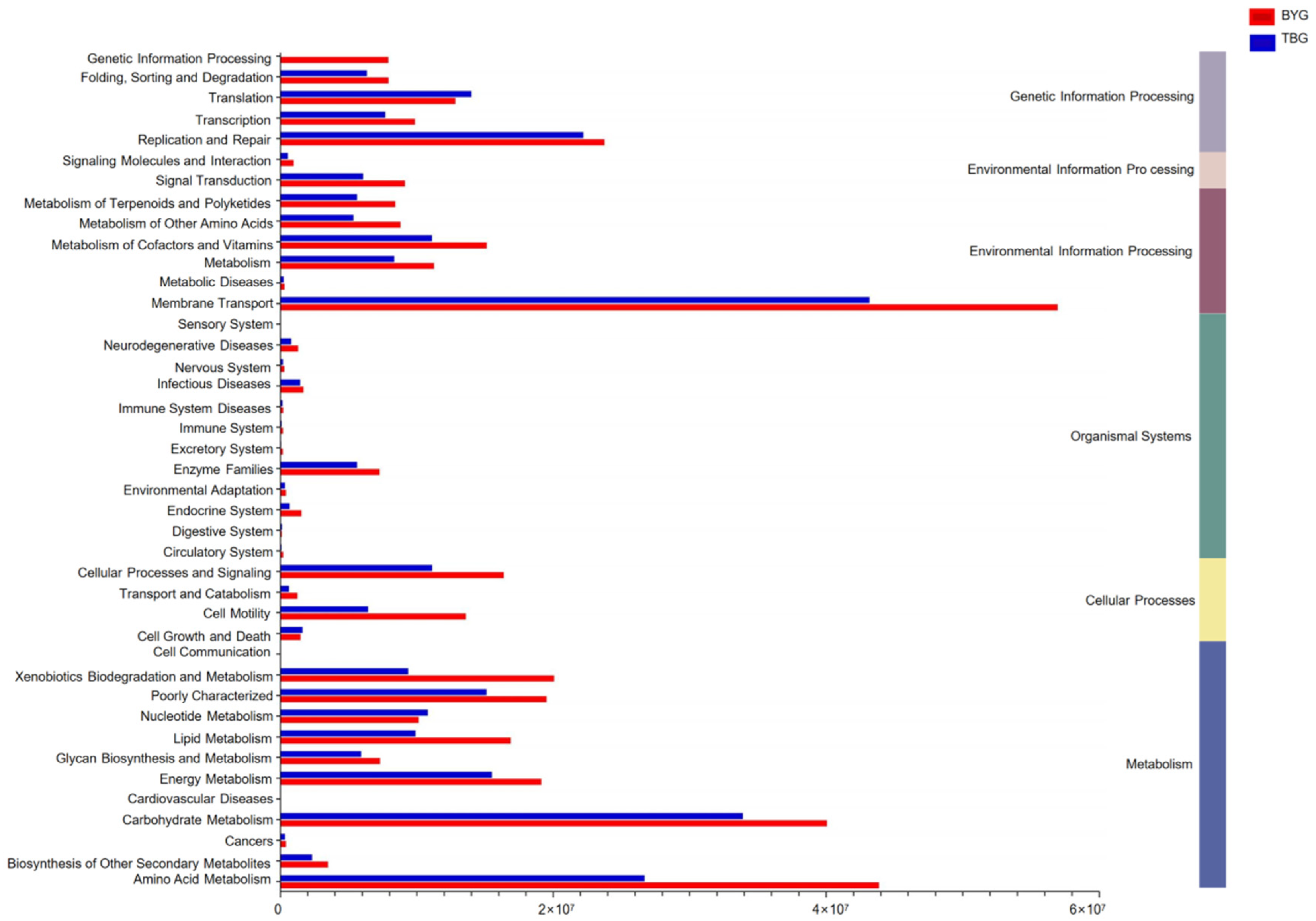
3.4. Pathway Analysis between Different Groups
3.5. Microstructure Analysis and the Food Comparison
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, E.T.; Svanback, R.; Bohannan, B.J.M. Microbiomes as Metacommunities: Understanding host-associated microbes through metacommunity ecology. Trends Ecol. Evol. 2018, 33, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M., Jr.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Song, P.; Lin, G.; Huang, Y.; Wang, L.; Zhou, X.; Li, S.; Zhang, T. Gut microbiota plasticity influences the adaptability of wild and domestic animals in co-inhabited areas. Front. Microbiol. 2020, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Sanders, J.G.; Delsuc, F.; Metcalf, J.; Amato, K.; Taylor, M.W.; Mazel, F.; Lutz, H.L.; Winker, K.; Graves, G.R.; et al. Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. mBio 2020, 11, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Beauchamp, G.; Li, Z. Coordination and synchronisation of anti-predation vigilance in two crane species. PLoS ONE 2011, 6, e26447. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Luo, W.; Moller, A.P.; Zhang, Y.; Yang, X. Vigilance strategy differentiation between sympatric threatened and common crane species. Behav. Processes 2020, 176, 104119. [Google Scholar] [CrossRef] [PubMed]
- Jarma, D.; Sanchez, M.I.; Green, A.J.; Peralta-Sanchez, J.M.; Hortas, F.; Sanchez-Melsio, A.; Borrego, C.M. Faecal microbiota and antibiotic resistance genes in migratory waterbirds with contrasting habitat use. Sci. Total Environ. 2021, 783, 146872. [Google Scholar] [CrossRef]
- Luo, J.; Wang, Y.; Gao, Z.; Wang, W. The excessive enrichment of trace elements in migratory and breeding red-crowned cranes (Grus japonensis) in China. Environ. Sci. Pollut. Res. Int. 2017, 24, 16351–16363. [Google Scholar] [CrossRef]
- Gu, J.; Zhou, L. Intestinal microbes of hooded cranes (Grus monacha) wintering in three lakes of the middle and lower Yangtze River Floodplain. Animals 2021, 11, 1390. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, C.; Arroyo, J.; Even, M.; Cauquil, L.; Pascal, G.; Fernandez, X.; Lavigne, F.; Davail, S.; Combes, S.; Ricaud, K. The intestinal microbial composition in Greylag geese differs with steatosis induction mode: Spontaneous or induced by overfeeding. Anim. Microbiome 2021, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, Y.; Cao, L.; Yin, H.; Xu, M.; Wang, Z.; Liu, Y.; Wang, X.; Deng, Y. Habitat environments impacted the gut microbiome of long-distance migratory swan geese but central species conserved. Sci. Rep. 2018, 8, 13314. [Google Scholar] [CrossRef] [PubMed]
- Videvall, E.; Strandh, M.; Engelbrecht, A.; Cloete, S.; Cornwallis, C.K. Measuring the gut microbiome in birds: Comparison of faecal and cloacal sampling. Mol. Ecol. Resour. 2018, 18, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.S.; Pardal, S.; Proenca, D.N.; Lopes, R.J.; Ramos, J.A.; Mendes, L.; Morais, P.V. Diversity of cloacal microbial community in migratory shorebirds that use the Tagus estuary as stopover habitat and their potential to harbor and disperse pathogenic microorganisms. FEMS Microbiol. Ecol. 2012, 82, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Zhang, S.; Xu, H.; Kong, F.; Yu, X.; Wang, P.; Yang, M.; Li, D.; Zhang, M.; Ni, Q.; et al. Gut microbiota of Tibetans and Tibetan pigs varies between high and low altitude environments. Microbiol. Res. 2020, 235, 126447. [Google Scholar] [CrossRef]
- Hird, S.M.; Sanchez, C.; Carstens, B.C.; Brumfield, R.T. Comparative gut microbiota of 59 neotropical bird species. Front. Microbiol. 2015, 6, 1403. [Google Scholar] [CrossRef]
- Alberdi, A.; Aizpurua, O.; Bohmann, K.; Zepeda-Mendoza, M.L.; Gilbert, M.T.P. Do vertebrate gut metagenomes confer rapid ecological adaptation? Trends Ecol. Evol. 2016, 31, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.B.; Moore, F.R.; Wang, S.A. Characterization of the gut microbiota of migratory passerines during stopover along the northern coast of the Gulf of Mexico. J. Avian Biol. 2016, 47, 659–668. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Koskey, A.M.; Fisher, J.C.; Traudt, M.F.; Newton, R.J.; McLellan, S.L. Analysis of the gull fecal microbial community reveals the dominance of Catellicoccus marimammalium in relation to culturable Enterococci. Appl. Environ. Microbiol. 2014, 80, 757–765. [Google Scholar] [CrossRef]
- Parfrey, L.W.; Walters, W.A.; Lauber, C.L.; Clemente, J.C.; Berg-Lyons, D.; Teiling, C.; Kodira, C.; Mohiuddin, M.; Brunelle, J.; Driscoll, M.; et al. Communities of microbial eukaryotes in the mammalian gut within the context of environmental eukaryotic diversity. Front. Microbiol. 2014, 5, 298. [Google Scholar] [CrossRef]
- Waite, D.W.; Taylor, M.W. Exploring the avian gut microbiota: Current trends and future directions. Front. Microbiol. 2015, 6, 673. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.; Mourkas, E.; Gonzalez-Acuna, D.; Olsen, B.; Ellstrom, P. Evaluation and optimization of microbial DNA extraction from fecal samples of wild Antarctic bird species. Infect. Ecol. Epidemiol. 2017, 7, 1386536. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 1. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.Y.; Xia, J.G. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Zhang, N.; Zhou, L.; Yang, Z.; Gu, J. Effects of food changes on intestinal bacterial diversity of wintering hooded cranes (Grus monacha). Animals 2021, 11, 433. [Google Scholar] [CrossRef]
- Palagi, E.; Bergman, T.J. Bridging captive and wild studies: Behavioral plasticity and social complexity in Theropithecus gelada. Animals 2021, 11, 3003. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Willemsen, D.; Popkes, M.; Metge, F.; Gandiwa, E.; Reichard, M.; Valenzano, D.R. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. eLife 2017, 6, e27014. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.B.; Al-Ghalith, G.A.; Long, H.T.; Tuan, B.V.; Cabana, F.; Huang, H.; Vangay, P.; Ward, T.; Minh, V.V.; Tam, N.A.; et al. Associations between nutrition, gut microbiome, and health in a novel nonhuman primate model. Sci. Rep. 2018, 8, 11159. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.; Safdar, N. Fecal microbiota transplantation for the treatment of Clostridium difficile infection. J. Hosp. Med. 2016, 11, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.H. Studies on fusobacteria associated with periodontal diseases. Aust. Dent. J. 1998, 43, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front. Cell Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, T.; Beasley, D.E.; Hedenec, P.; Xiao, Z.; Zhang, S.; Li, J.; Lin, Q.; Li, X. Diet diversity is associated with beta but not alpha diversity of pika gut microbiota. Front. Microbiol. 2016, 7, 1169. [Google Scholar] [CrossRef] [PubMed]
- Riva, A.; Borgo, F.; Lassandro, C.; Verduci, E.; Morace, G.; Borghi, E.; Berry, D. Pediatric obesity is associated with an altered gut microbiota and discordant shifts in Firmicutes populations. Environ. Microbiol. 2017, 19, 95–105. [Google Scholar] [CrossRef]
- Medawar, E.; Huhn, S.; Villringer, A.; Veronica Witte, A. The effects of plant-based diets on the body and the brain: A systematic review. Transl. Psychiatry 2019, 9, 226. [Google Scholar] [CrossRef]
- Cani, P.D.; Delzenne, N.M. The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J.; Pazienza, V. Impact of different types of diet on gut microbiota profiles and cancer prevention and treatment. Medicina 2019, 55, 84. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Fan, X.; Wang, Y.; Kusstatscher, P.; Duan, J.; Wu, S.; Chen, S.; Qiao, K.; Wang, Y.; Ma, B.; et al. Bacterial seed endophyte shapes disease resistance in rice. Nat. Plants 2021, 7, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Kunakom, S.; Eustaquio, A.S. Burkholderia as a source of natural products. J. Nat. Prod. 2019, 82, 2018–2037. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhai, Q.; Zhang, H.; Chen, W.; Hill, C. Gut colonization mechanisms of Lactobacillus and Bifidobacterium: An argument for personalized designs. Annu. Rev. Food Sci. Technol. 2021, 12, 213–233. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microb. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Sobs | Simpsoneven | Chao1 | Shannon | Ace | Coverage |
|---|---|---|---|---|---|---|
| BYG1 | 284 | 0.005385 | 316.2826 | 1.174740 | 324.2621 | 0.999194 |
| BYG2 | 1333 | 0.005165 | 1375.0620 | 4.296252 | 1374.3710 | 0.998564 |
| BYG3 | 863 | 0.001994 | 950.4793 | 1.786709 | 845.7505 | 0.997861 |
| BYG4 | 1104 | 0.002353 | 1191.3440 | 2.767240 | 1168.6470 | 0.998095 |
| BYG5 | 461 | 0.003055 | 580.1563 | 1.076843 | 560.8193 | 0.998183 |
| BYG6 | 739 | 0.002397 | 939.1724 | 1.465221 | 926.8143 | 0.996835 |
| TBG1 | 621 | 0.005376 | 714.4304 | 2.358204 | 707.4409 | 0.998212 |
| TBG2 | 546 | 0.003534 | 705.0723 | 1.414751 | 687.7625 | 0.997612 |
| TBG3 | 280 | 0.006499 | 323.3636 | 1.497142 | 323.6567 | 0.999209 |
| TBG4 | 139 | 0.007739 | 229.2308 | 0.233573 | 324.2930 | 0.998989 |
| TBG5 | 528 | 0.012882 | 602.6522 | 2.817375 | 609.5538 | 0.998505 |
| TGB6 | 173 | 0.024830 | 285.8571 | 2.007119 | 394.1489 | 0.998828 |
| TBG7 | 766 | 0.056894 | 849.7391 | 4.664402 | 839.9976 | 0.998418 |
| TBG8 | 196 | 0.006157 | 328.0000 | 0.529410 | 410.3215 | 0.998711 |
| TBG9 | 190 | 0.011155 | 293.0526 | 1.309319 | 422.3113 | 0.998696 |
| TBG10 | 174 | 0.010918 | 384.0370 | 0.919415 | 634.3366 | 0.998432 |
| TBG11 | 295 | 0.006109 | 427.2222 | 0.892827 | 524.5832 | 0.998242 |
| TBG12 | 428 | 0.008311 | 533.0196 | 2.051407 | 523.7572 | 0.998476 |
| TBG13 | 268 | 0.007102 | 351.1429 | 1.047081 | 443.1174 | 0.998579 |
| TBG14 | 544 | 0.003021 | 636.6389 | 1.465143 | 642.9121 | 0.998300 |
| TBG15 | 350 | 0.008212 | 467.2979 | 1.368138 | 511.8669 | 0.997817 |
| Family | Species | TBG | BYG | ||||
|---|---|---|---|---|---|---|---|
| F (Times) | D (%) | RD (%) | F (Times) | D (%) | RD (%) | ||
| Gramineae | Triticum aestivum | 268 | 50.13 | 50.05 | |||
| Zea mays | 47 | 8.77 | 8.76 | 151 | 24.31 | 20.92 | |
| Phragmites australis | 25 | 4.67 | 4.66 | 130 | 20.92 | 18.01 | |
| Malvaceae | Abutilontheophrasti | 24 | 4.48 | 4.47 | |||
| Cyperaceae | Carexbreviculmis | 34 | 6.35 | 6.34 | |||
| Rosaceae | Potentilla chinensis | 53 | 9.89 | 9.88 | 63 | 26.24 | 22.58 |
| Leguminous | Glycine max | 31 | 5.79 | 5.78 | 132 | 21.24 | 18.28 |
| Ranunculaceae | Ranunculus japonicus | 7 | 1.31 | 1.30 | |||
| Composite | Artemisia capillaris | 8 | 1.49 | 1.49 | |||
| Polygonaceae | Polygonum criopolitanum | 42 | 6.75 | 5.81 | |||
| Cruciferae | Capsella bursa | 68 | 10.93 | 9.41 | |||
| Araceae | Acorus tatarinowii | 15 | 2.41 | 2.08 | |||
| Others | 39 | 7.28 | 7.27 | 21 | 3.38 | 2.91 | |
| Plant | R | p-Value |
|---|---|---|
| Zea mays | 0.43470 | 0.003 |
| Glycine max | 0.53895 | 0.006 |
| Phragmites australia | 0.70953 | 0.005 |
| Potentilla amurensis | 0.13408 | 0.331 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Wu, N.; Liu, X.; Zhang, L.; Zhao, D. Diet Drives Gut Bacterial Diversity of Wild and Semi-Captive Common Cranes (Grus grus). Animals 2024, 14, 1566. https://doi.org/10.3390/ani14111566
Wu H, Wu N, Liu X, Zhang L, Zhao D. Diet Drives Gut Bacterial Diversity of Wild and Semi-Captive Common Cranes (Grus grus). Animals. 2024; 14(11):1566. https://doi.org/10.3390/ani14111566
Chicago/Turabian StyleWu, Hong, Nan Wu, Xinchen Liu, Lei Zhang, and Dapeng Zhao. 2024. "Diet Drives Gut Bacterial Diversity of Wild and Semi-Captive Common Cranes (Grus grus)" Animals 14, no. 11: 1566. https://doi.org/10.3390/ani14111566