
Influence of Varied Environment Conditions on the Gut Microbiota of Yaks

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and 16S rDNA Amplicon Sequencing
2.3. Bioinformatics and Statistical Analysis
2.4. Statistical Analysis
3. Results
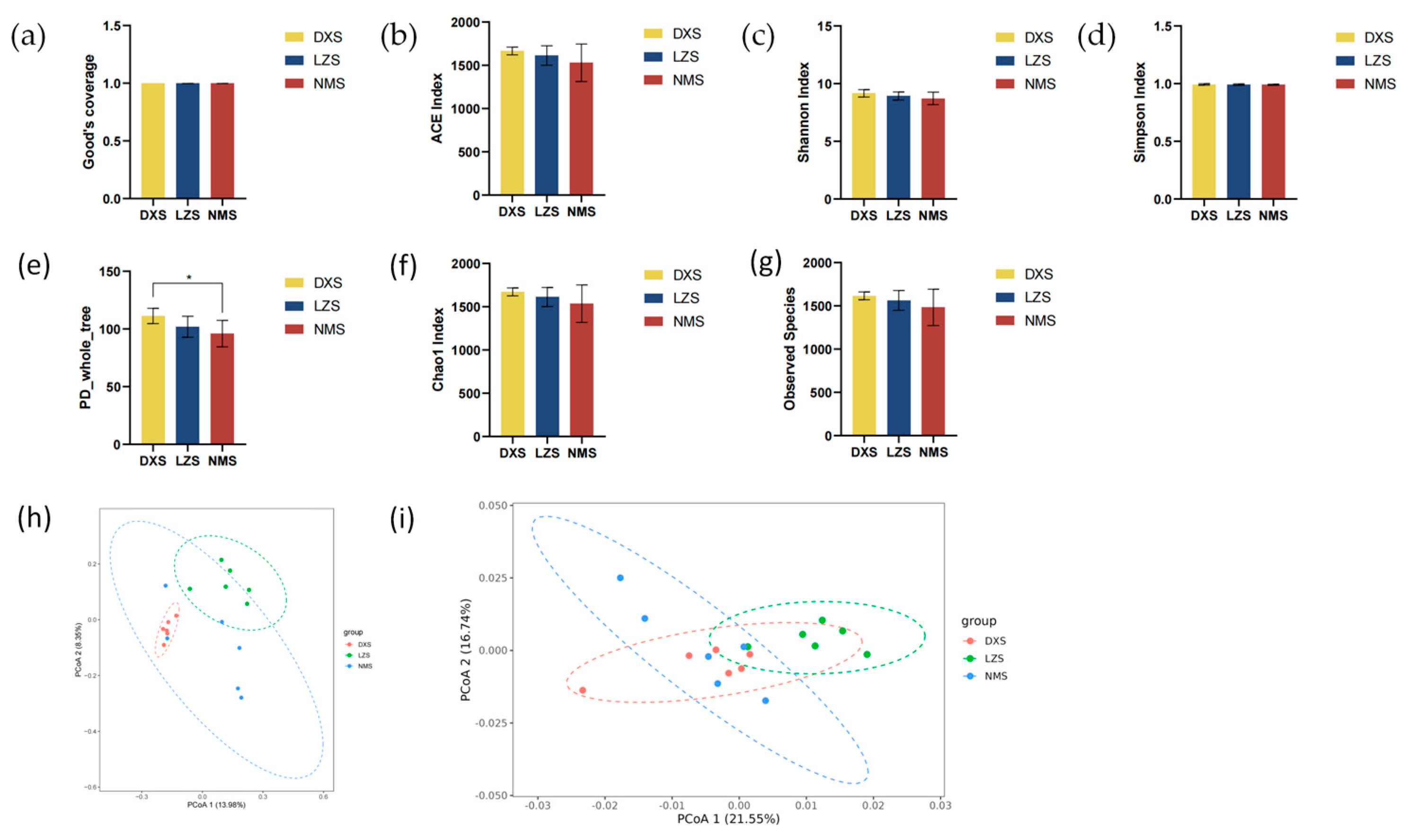
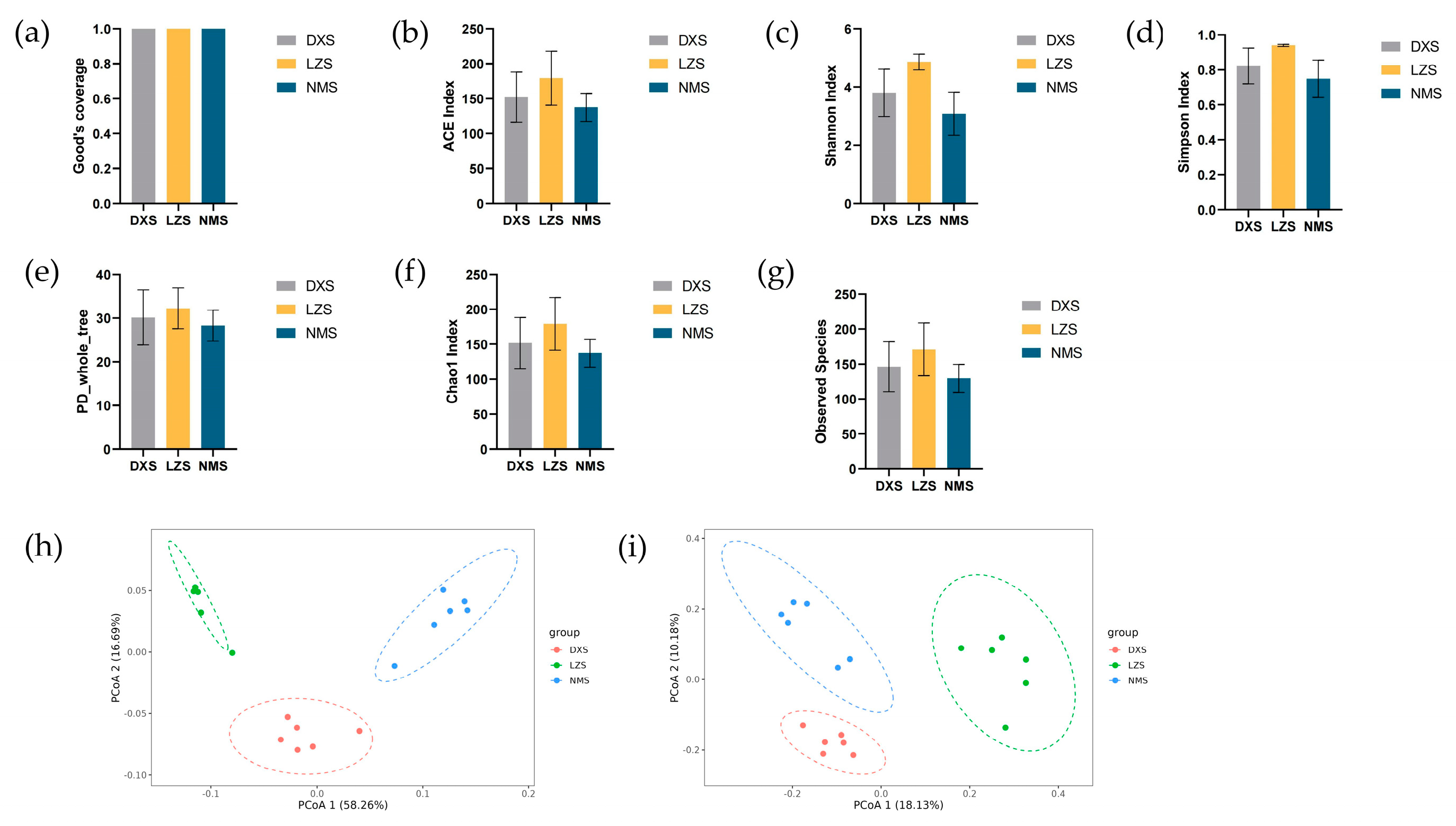
3.1. Microbial Diversity
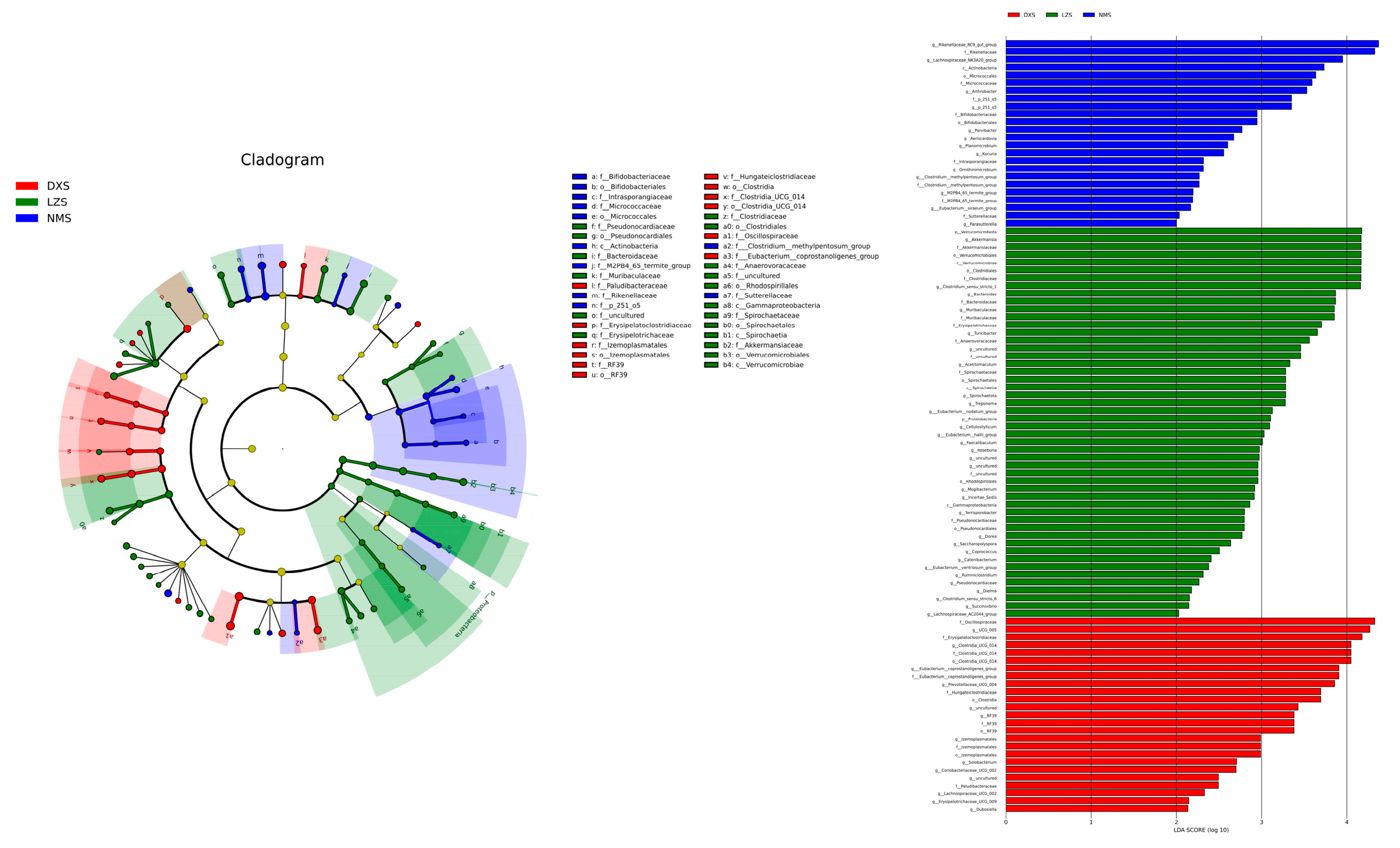
3.2. Intestinal Bacterial Composition Analysis
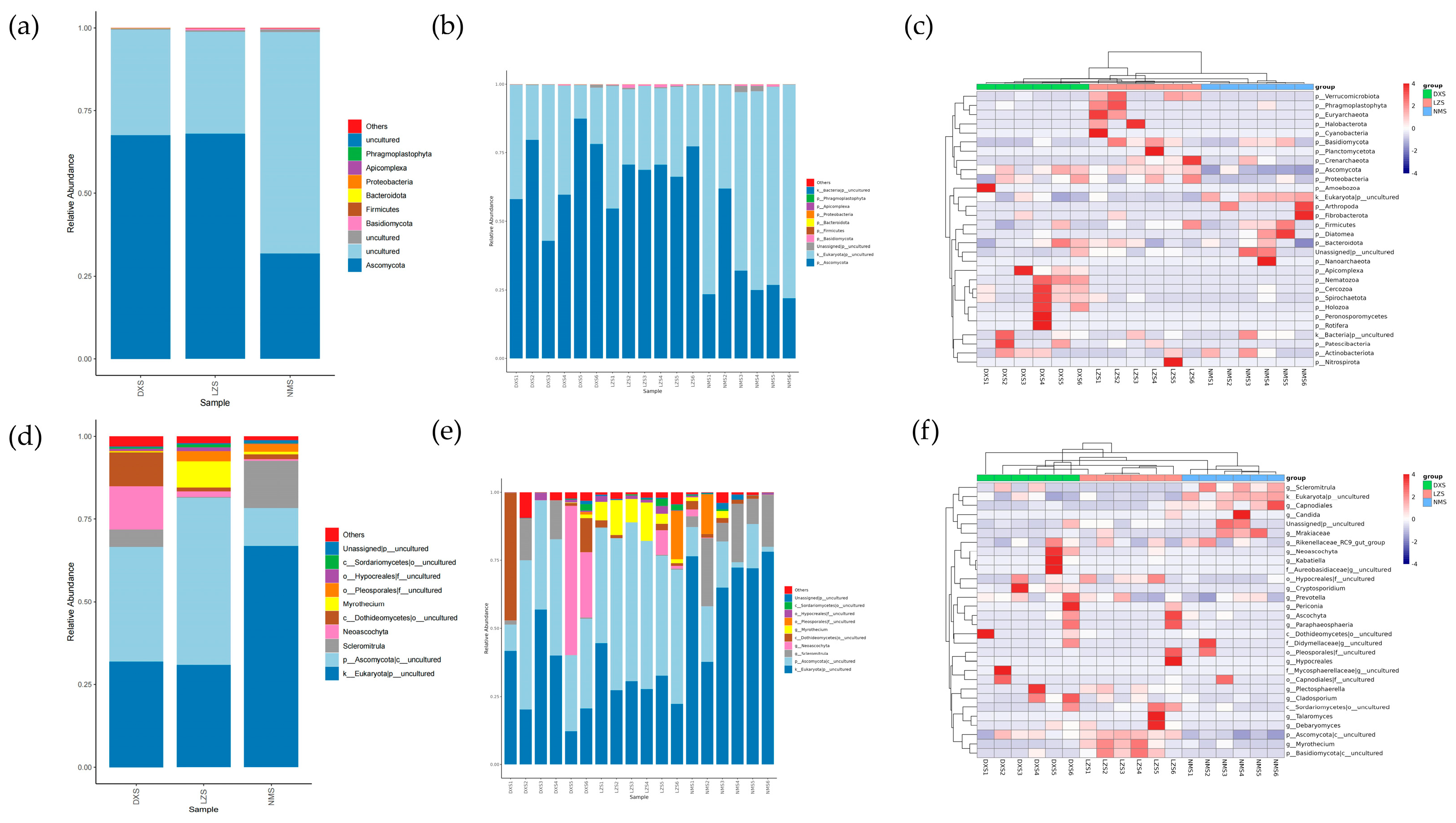
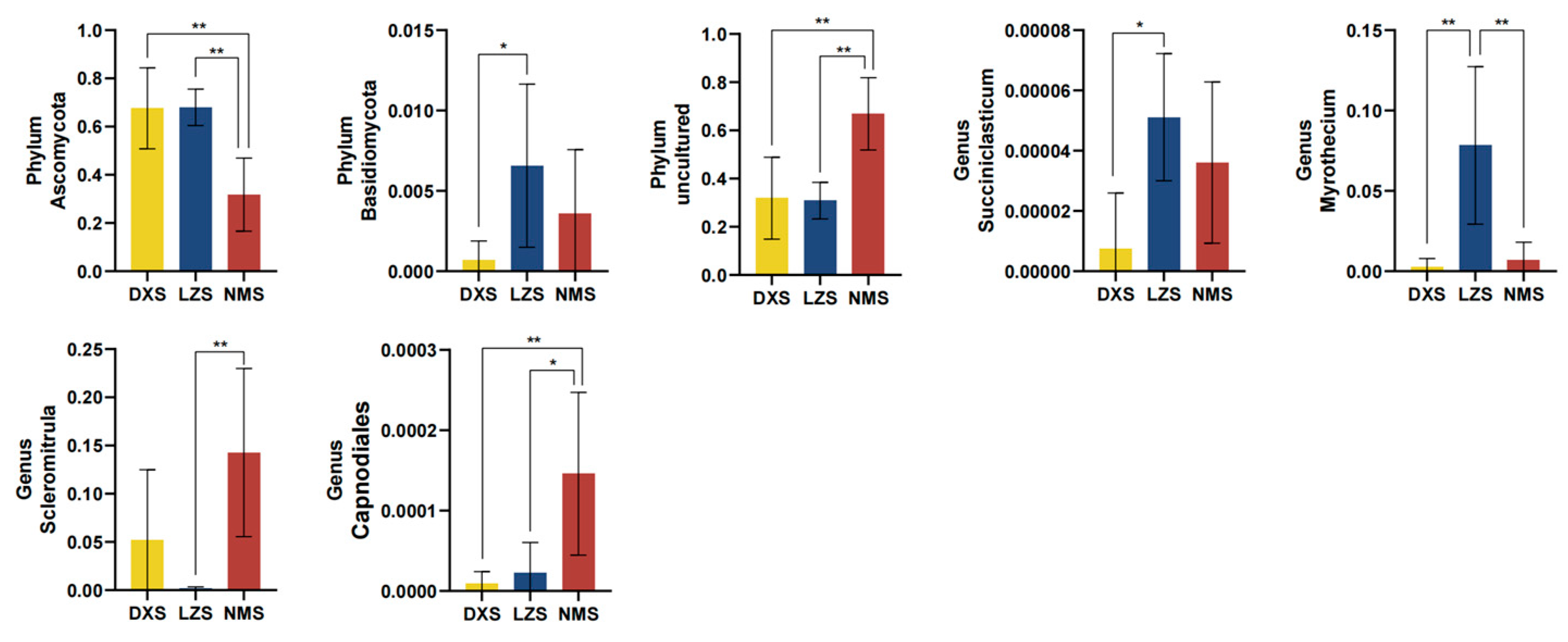
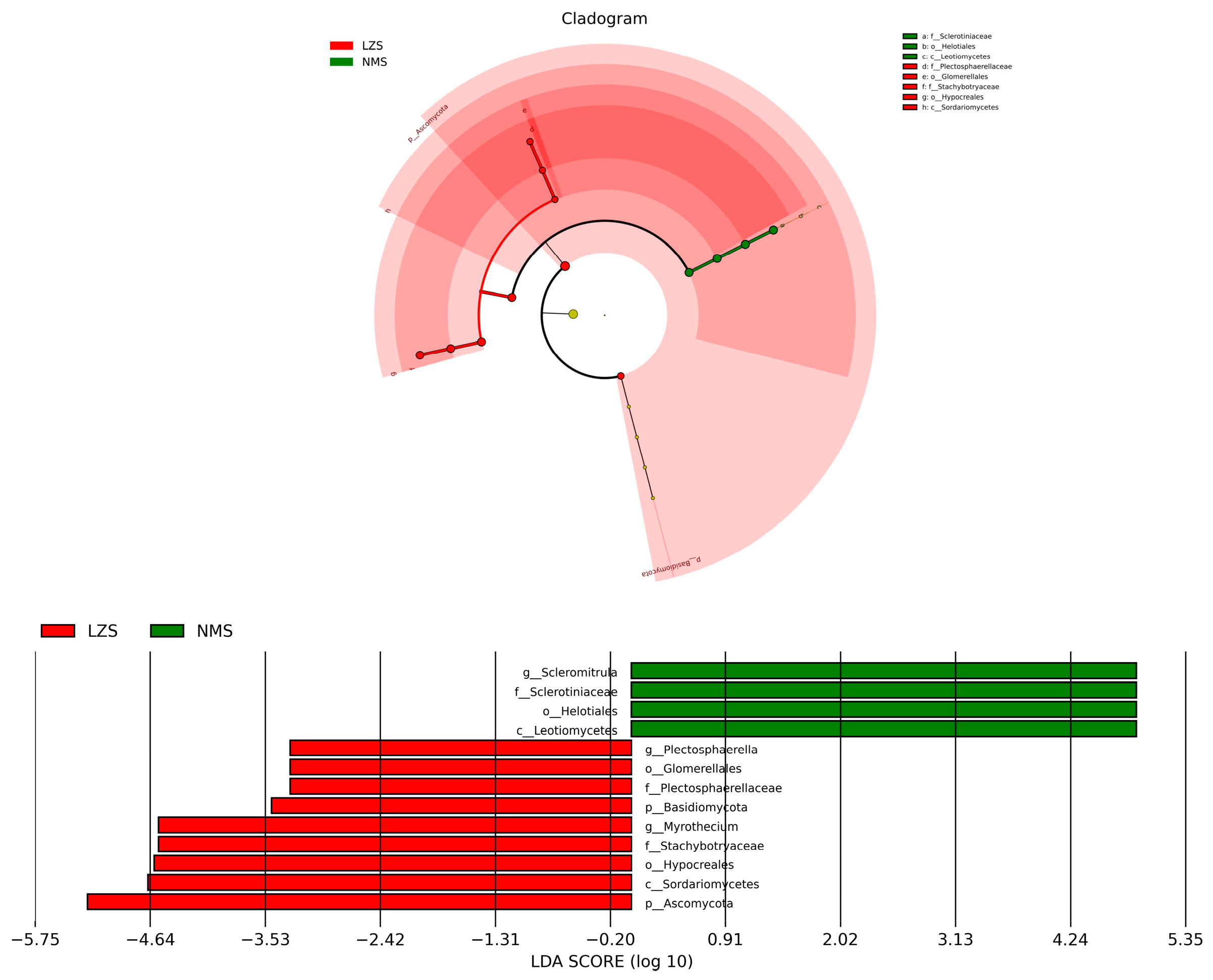
3.3. Intestinal Fungal Composition Analysis
3.4. Metabolic Function Prediction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El-Sayed, A.; Aleya, L.; Kamel, M. Microbiota’s role in health and diseases. Env. Sci. Pollut. Res. Int. 2021, 28, 36967–36983. [Google Scholar] [CrossRef]
- Perlman, D.; Martínez-Álvaro, M.; Moraïs, S.; Altshuler, I.; Hagen, L.H.; Jami, E.; Roehe, R.; Pope, P.B.; Mizrahi, I. Concepts and Consequences of a Core Gut Microbiota for Animal Growth and Development. Annu. Rev. Anim. Biosci. 2022, 10, 177–201. [Google Scholar] [CrossRef]
- Coman, V.; Vodnar, D.C. Gut microbiota and old age: Modulating factors and interventions for healthy longevity. Exp. Gerontol. 2020, 141, 111095. [Google Scholar] [CrossRef]
- Zhang, F.; Xue, H.; Dong, X.; Li, M.; Zheng, X.; Li, Z.; Xu, J.; Wang, W.; Wei, C. Long-read sequencing of 111 rice genomes reveals significantly larger pan-genomes. Genome Res. 2022, 32, 853–863. [Google Scholar] [CrossRef]
- AAdak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell. Mol. Life Sci. 2019, 76, 473–493. [Google Scholar] [CrossRef]
- Chen, L.; Wang, J. Gut microbiota and inflammatory bowel disease. WIREs Mech. Dis. 2022, 14, e1540. [Google Scholar] [CrossRef]
- Sittipo, P.; Lobionda, S.; Lee, Y.K.; Maynard, C.L. Intestinal microbiota and the immune system in metabolic diseases. J. Microbiol. 2018, 56, 154–162. [Google Scholar] [CrossRef]
- Kayama, H.; Okumura, R.; Takeda, K. Interaction Between the Microbiota, Epithelia, and Immune Cells in the Intestine. Annu. Rev. Immunol. 2020, 38, 23–48. [Google Scholar] [CrossRef]
- Welch, C.B.; Ryman, V.E.; Pringle, T.D.; Lourenco, J.M. Utilizing the Gastrointestinal Microbiota to Modulate Cattle Health through the Microbiome-Gut-Organ Axes. Microorganisms 2022, 10, 1391. [Google Scholar] [CrossRef]
- O’Hara, E.; Neves, A.L.A.; Song, Y.; Guan, L.L. The Role of the Gut Microbiome in Cattle Production and Health: Driver or Passenger? Annu. Rev. Anim. Biosci. 2020, 8, 199–220. [Google Scholar] [CrossRef]
- Arshad, M.A.; Hassan, F.U.; Rehman, M.S.; Huws, S.A.; Cheng, Y.; Din, A.U. Gut microbiome colonization and development in neonatal ruminants: Strategies, prospects, and opportunities. Anim. Nutr. 2021, 7, 883–895. [Google Scholar] [CrossRef]
- Bai, X.; Liu, G.; Yang, J.; Duan, Y.; Zhu, J.; Li, X.Q.C. The effect of high-altitude hypoxia on drug metabolism is mediated by gut microbiota. Yaoxue Xuebao 2021, 56, 2787–2796. [Google Scholar]
- Fan, Q.; Wanapat, M.; Hou, F. Mineral Nutritional Status of Yaks (Bos Grunniens) Grazing on the Qinghai-Tibetan Plateau. Animals 2019, 9, 468. [Google Scholar] [CrossRef]
- Fan, Q.; Wanapat, M.; Yan, T.; Hou, F. Altitude influences microbial diversity and herbage fermentation in the rumen of yaks. BMC Microbiol. 2020, 20, 370. [Google Scholar] [CrossRef]
- Jia, H.F.; Mou, Y.M.; Danzeng, L.B.; Li, Y. Age investigation and growth history analysis of old trees in Nyemo County of Tibet, China. Ying Yong Sheng Tai Xue Bao 2018, 29, 2401–2410. [Google Scholar]
- Zhu, G.Q.; Yan, H.B.; Li, L.; Ohiolei, J.A.; Wu, Y.T.; Li, W.H.; Zhang, N.Z.; Fu, B.Q.; Jia, W.Z. First report on the phylogenetic relationship, genetic variation of Echinococcus shiquicus isolates in Tibet Autonomous Region, China. Parasit. Vectors 2020, 13, 590. [Google Scholar] [CrossRef]
- Li, B.; Jia, G.; Wen, D.; Zhao, X.; Zhang, J.; Xu, Q.; Zhao, X.; Jiang, N.; Liu, Z.; Wang, Y. Rumen microbiota of indigenous and introduced ruminants and their adaptation to the Qinghai-Tibetan plateau. Front. Microbiol. 2022, 13, 1027138. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, C.; Guo, T.; Wang, F.; Zeng, Y.; Ding, X.; Lu, Z.; Renqing, D.; Zhang, H.; Xu, X.; et al. Genetic signatures of high-altitude adaptation and geographic distribution in Tibetan sheep. Sci. Rep. 2020, 10, 18332. [Google Scholar] [CrossRef]
- Jing, X.; Ding, L.; Zhou, J.; Huang, X.; Degen, A.; Long, R. The adaptive strategies of yaks to live in the Asian highlands. Anim. Nutr. 2022, 9, 249–258. [Google Scholar] [CrossRef]
- Peng, M.S.; Liu, Y.H.; Shen, Q.K.; Zhang, X.H.; Dong, J.; Li, J.X.; Zhao, H.; Zhang, H.; Zhang, X.; He, Y.; et al. Genetic and cultural adaptations underlie the establishment of dairy pastoralism in the Tibetan Plateau. BMC Biol. 2023, 21, 208. [Google Scholar] [CrossRef]
- Shah, A.M.; Bano, I.; Qazi, I.H.; Matra, M.; Wanapat, M. “The Yak”-A remarkable animal living in a harsh environment: An overview of its feeding, growth, production performance, and contribution to food security. Front. Vet. Sci. 2023, 10, 1086985. [Google Scholar] [CrossRef]
- Nambu, R.; Warner, N.; Mulder, D.J.; Kotlarz, D.; McGovern, D.; Cho, J.; Klein, C.; Snapper, S.B.; Griffiths, A.M.; Iwama, I.; et al. A Systematic Review of Monogenic Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2022, 20, e653–e663. [Google Scholar] [CrossRef]
- Chen, L.; Qiu, Q.; Jiang, Y.; Wang, K.; Lin, Z.; Li, Z.; Bibi, F.; Yang, Y.; Wang, J.; Nie, W.; et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 2019, 364, eaav6202. [Google Scholar] [CrossRef]
- Xin, J.; Chai, Z.; Zhang, C.; Zhang, Q.; Zhu, Y.; Cao, H.; Zhong, J.; Ji, Q. Comparing the Microbial Community in Four Stomach of Dairy Cattle, Yellow Cattle and Three Yak Herds in Qinghai-Tibetan Plateau. Front. Microbiol. 2019, 10, 1547. [Google Scholar] [CrossRef]
- Huang, X.; Mi, J.; Denman, S.E.; Basangwangdui Pingcuozhandui Zhang, Q.; Long, R. McSweeney CS. Changes in rumen microbial community composition in yak in response to seasonal variations. J. Appl. Microbiol. 2022, 132, 1652–1665. [Google Scholar] [CrossRef]
- Yu, H.J.; Jing, C.; Xiao, N.; Zang, X.M.; Zhang, C.Y.; Zhang, X.; Qu, Y.N.; Li, Y.; Tan, Q.W. Structural difference analysis of adult’s intestinal flora basing on the 16S rDNA gene sequencing technology. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 12983–12992. [Google Scholar]
- Zhu, Q.; Wang, N.; Duan, B.; Wang, Q.; Wang, Y. Rhizosphere bacterial and fungal communities succession patterns related to growth of poplar fine roots. Sci. Total Environ. 2021, 756, 143839. [Google Scholar] [CrossRef]
- Moreno, L.A.; Cox, K.L. Quantification of dsDNA using the Hitachi F-7000 Fluorescence Spectrophotometer and PicoGreen dye. J. Vis. Exp. 2010, 45, e2465. [Google Scholar]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010, Pdb.prot5448. [Google Scholar] [CrossRef]
- Maki, K.A.; Wolff, B.; Varuzza, L.; Green, S.J.; Barb, J.J. Multi-amplicon microbiome data analysis pipelines for mixed orientation sequences using QIIME2: Assessing reference database, variable region and pre-processing bias in classification of mock bacterial community samples. PLoS ONE 2023, 18, e0280293. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory, C.J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega, T.R.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Takiishi, T.; Fenero, C.; Câmara, N. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017, 5, e1373208. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Tanaka, T.; Murata, S.; Miyabe, A.; Ishige, T.; Kawasaki, K.; Yokoyama, M.; Hashimoto, N.; Yamagata, K.; Nagano, H.; et al. Extension of bacterial rDNA sequencing for simultaneous methylation detection and its application in microflora analysis. Sci. Rep. 2023, 13, 5731. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.Y.; Robertson, I.D.; Ji, Q.M.; Dawa, Y.L.; Bruce, M. Evaluation of the economic impact of brucellosis in domestic yaks of Tibet. Transbound. Emerg. Dis. 2019, 66, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Bai, J.; Xu, D.; Li, F.; Zhang, Y.; Guo, X. Microbial Community Dynamics and Natural Fermentation Profiles of Ensiled Alpine Grass Elymus nutans Prepared From Different Regions of the Qinghai-Tibetan Plateau. Front. Microbiol. 2020, 11, 855. [Google Scholar] [CrossRef]
- Liu, M.; Li, T.; Yang, J.; Li, S.S.; Yu, Z.J.; Liu, J.Z. Seasonal abundance and activity of the tick Dermacentor everestianus (Acari: Ixodidae) in the Tibetan Plateau, China. Exp. Appl. Acarol. 2020, 81, 609–619. [Google Scholar] [CrossRef]
- Ma, Y.; Ma, S.; Chang, L.; Wang, H.; Ga, Q.; Ma, L.; Bai, Z.; Shen, Y.; Ge, R.L. Gut microbiota adaptation to high altitude in indigenous animals. Biochem. Biophys. Res. Commun. 2019, 516, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.A.; Martins, F.M.; Nachman, M.W. Altitudinal variation of the gut microbiota in wild house mice. Mol. Ecol. 2019, 28, 2378–2390. [Google Scholar] [CrossRef] [PubMed]
- Adak, A.; Maity, C.; Ghosh, K.; Pati, B.R.; Mondal, K.C. Dynamics of predominant microbiota in the human gastrointestinal tract and change in luminal enzymes and immunoglobulin profile during high-altitude adaptation. Folia Microbiol. 2013, 58, 523–528. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Deng, J.; Liu, J.; Fu, J.; Xiong, H.; Luo, W.; Xiong, J. Seasonal variations in the composition and diversity of gut microbiota in white-lipped deer (Cervus albirostris). PeerJ 2022, 10, e13753. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Gao, H.; Qin, W.; Song, P.; Wang, H.; Zhang, J.; Liu, D.; Wang, D.; Zhang, T. Marked Seasonal Variation in Structure and Function of Gut Microbiota in Forest and Alpine Musk Deer. Front. Microbiol. 2021, 12, 699797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shi, M.; Fan, M.; Xu, S.; Li, Y.; Zhang, T.; Cha, M.; Liu, Y.; Guo, X.; Chen, Q.; et al. Comparative Analysis of Gut Microbiota Changes in Père David’s Deer Populations in Beijing Milu Park and Shishou, Hubei Province in China. Front. Microbiol. 2018, 9, 1258. [Google Scholar] [CrossRef] [PubMed]
- Walters, K.E.; Martiny, J. Alpha-, beta-, and gamma-diversity of bacteria varies across habitats. PLoS ONE 2020, 15, e0233872. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, X.; Li, A.; Waqas, M.; Gao, X.; Li, K.; Xie, G.; Zhang, J.; Mehmood, K.; Zhao, S.; et al. Characterization of the microbial community structure in intestinal segments of yak (Bos grunniens). Anaerobe 2020, 61, 102115. [Google Scholar] [CrossRef] [PubMed]
- Aricha, H.; Simujide, H.; Wang, C.; Zhang, J.; Lv, W.; Jimisi, X.; Liu, B.; Chen, H.; Zhang, C.; He, L.; et al. Comparative Analysis of Fecal Microbiota of Grazing Mongolian Cattle from Different Regions in Inner Mongolia, China. Animals 2021, 11, 1938. [Google Scholar] [CrossRef]
- Li, A.; Yang, Y.; Qin, S.; Lv, S.; Jin, T.; Li, K.; Han, Z.; Li, Y. Microbiome analysis reveals gut microbiota alteration of early-weaned Yimeng black goats with the effect of milk replacer and age. Microb. Cell Fact. 2021, 20, 78. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, Y.; Wei, H.; Chen, Y.; Shang, H. Study on the Diversity and Function of Gut Microbiota in Pigs Following Long-Term Antibiotic and Antibiotic-Free Breeding. Curr. Microbiol. 2020, 77, 4114–4128. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Chen, T.; Green, S.J.; Mutlu, E.; Martin, B.R.; Rumpagaporn, P.; Patterson, J.A.; Keshavarzian, A.; Hamaker, B.R. Physical Inaccessibility of a Resistant Starch Shifts Mouse Gut Microbiota to Butyrogenic Firmicutes. Mol. Nutr. Food Res. 2019, 63, e1801012. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, J.; Wu, X.; Zhang, J.; Chen, Z.; Kang, X.; Lv, Z. The fate of anaerobic syntrophy in anaerobic digestion facing propionate and acetate accumulation. Waste Manag. 2021, 124, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Orellana, L.H.; Francis, T.B.; Ferraro, M.; Hehemann, J.H.; Fuchs, B.M.; Amann, R.I. Verrucomicrobiota are specialist consumers of sulfated methyl pentoses during diatom blooms. ISME J. 2022, 16, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef]
- Macchione, I.G.; Lopetuso, L.R.; Ianiro, G.; Napoli, M.; Gibiino, G.; Rizzatti, G.; Petito, V.; Gasbarrini, A.; Scaldaferri, F. Akkermansia muciniphila: Key player in metabolic and gastrointestinal disorders. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8075–8083. [Google Scholar]
- Bradley, P.H.; Pollard, K.S. Proteobacteria explain significant functional variability in the human gut microbiome. Microbiome 2017, 5, 36. [Google Scholar] [CrossRef]
- Do, K.H.; Ko, S.H.; Kim, K.B.; Seo, K.; Lee, W.K. Comparative Study of Intestinal Microbiome in Patients with Ulcerative Colitis and Healthy Controls in Korea. Microorganisms 2023, 11, 2750. [Google Scholar] [CrossRef]
- Sun, B.; Wang, X.; Bernstein, S.; Huffman, M.A.; Xia, D.P.; Gu, Z.; Chen, R.; Sheeran, L.K.; Wagner, R.S.; Li, J. Marked variation between winter and spring gut microbiota in free-ranging Tibetan Macaques (Macaca thibetana). Sci. Rep. 2016, 6, 26035. [Google Scholar] [CrossRef]
- Barrak, I.; Stájer, A.; Gajdács, M.; Urbán, E. Small, but smelly: The importance of Solobacterium moorei in halitosis and other human infections. Heliyon 2020, 6, e05371. [Google Scholar] [CrossRef] [PubMed]
- Sárvári, K.P.; Sántha, D.; Kovács, R.; Körmöndi, S.; Pető, Z.; Vereb, T.; Sztanó, B. Six cases of Solobacterium moorei isolated alone or in mixed culture in Hungary and comparison with previously published cases. Anaerobe 2020, 65, 102241. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.C.; Lian, C.A.; He, L.S. Genomic Characterization of a Novel Tenericutes Bacterium from Deep-Sea Holothurian Intestine. Microorganisms 2020, 8, 1874. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.B.; Gonzalez, E.L.; Choy, K.; Faull, K.F.; Jewell, T.; Arellano, A.; Liang, J.; Yu, K.B.; Paramo, J.; Hsiao, E.Y. Gut microbiota Turicibacter strains differentially modify bile acids and host lipids. Nat. Commun. 2023, 14, 3669. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Zeng, L.Y.; Zhao, Y.F.; Tang, H.X.; Lei, H.; Wan, Y.F.; Deng, Y.Q.; Liu, K.X. Causal effects of gut microbiota on sepsis: A two-sample Mendelian randomization study. Front. Microbiol. 2023, 14, 1167416. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Song, H.; Zhang, X.; Song, G.; Wang, Y.; Ding, X.; Duan, X.; Li, L.; Sun, T.; Kan, Q. Metformin attenuated sepsis-related liver injury by modulating gut microbiota. Emerg. Microbes Infect. 2022, 11, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.; Kong, J.Y.; Stothard, P.; Willing, B.P. Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 2019, 13, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.Z.; Wang, X.; Yan, X. Effects of Aeriscardovia aeriphila on growth performance, antioxidant functions, immune responses, and gut microbiota in broiler chickens. J. Zhejiang Univ. Sci. B 2023, 24, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Pacífico, C.; Petri, R.M.; Ricci, S.; Mickdam, E.; Wetzels, S.U.; Neubauer, V.; Zebeli, Q. Unveiling the Bovine Epimural Microbiota Composition and Putative Function. Microorganisms 2021, 9, 342. [Google Scholar] [CrossRef]
- van Gylswyk, N.O. Succiniclasticum ruminis gen. nov., sp. nov., a ruminal bacterium converting succinate to propionate as the sole energy-yielding mechanism. Int. J. Syst. Bacteriol. 1995, 45, 297–300. [Google Scholar] [CrossRef]
- de Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut microbiome and health: Mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Samaja, M.; Crespi, T.; Guazzi, M.; Vandegriff, K.D. Oxygen transport in blood at high altitude: Role of the hemoglobin-oxygen affinity and impact of the phenomena related to hemoglobin allosterism and red cell function. Eur. J. Appl. Physiol. 2003, 90, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tang, J.; Zhang, R.; Zhan, S.; Zhong, T.; Guo, J.; Wang, Y.; Cao, J.; Li, L.; Zhang, H.; et al. Cold exposure induces lipid dynamics and thermogenesis in brown adipose tissue of goats. BMC Genom. 2022, 23, 528. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, H.; Xu, T.; Hu, Y.; Jiang, Y. Acute exposure to simulated high-altitude hypoxia alters gut microbiota in mice. Arch. Microbiol. 2022, 204, 412. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Garcia, J.A.; Xiang, Y.; De Veylder, L. Cell cycle checkpoint control in response to DNA damage by environmental stresses. Plant J. 2022, 109, 490–507. [Google Scholar] [CrossRef]
- Ferrero-Serrano, Á.; Sylvia, M.M.; Forstmeier, P.C.; Olson, A.J.; Ware, D.; Bevilacqua, P.C.; Assmann, S.M. Experimental demonstration and pan-structurome prediction of climate-associated riboSNitches in Arabidopsis. Genome Biol. 2022, 23, 101. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Input | Filtered | Effectie (%) | AvgLen (bp) | GC (%) |
|---|---|---|---|---|---|
| DXS1 | 149055 | 142514 | 95.61 | 226.85 | 53.405 |
| DXS2 | 141336 | 135362 | 95.77 | 226.4 | 53.23 |
| DXS3 | 145569 | 139397 | 95.76 | 225.95 | 53.295 |
| DXS4 | 145326 | 139083 | 95.7 | 227.35 | 53.285 |
| DXS5 | 135439 | 129753 | 95.8 | 226.95 | 53.735 |
| DXS6 | 140421 | 134845 | 96.03 | 226.3 | 53.725 |
| LZS1 | 148314 | 142163 | 95.85 | 226.8 | 53.44 |
| LZS2 | 135054 | 129488 | 95.88 | 226.4 | 52.755 |
| LZS3 | 139428 | 133871 | 96.01 | 225.8 | 53.24 |
| LZS4 | 144026 | 138466 | 96.14 | 227.4 | 53.18 |
| LZS5 | 142422 | 136824 | 96.07 | 226.85 | 53.33 |
| LZS6 | 136476 | 131350 | 96.24 | 226.3 | 53.655 |
| NMS1 | 142805 | 136477 | 95.57 | 225.85 | 53.59 |
| NMS2 | 141404 | 135608 | 95.9 | 227.45 | 53.67 |
| NMS3 | 143945 | 137735 | 95.69 | 226.95 | 53.53 |
| NMS4 | 136989 | 131526 | 96.01 | 226.3 | 52.86 |
| NMS5 | 139503 | 133409 | 95.63 | 225.85 | 52.98 |
| NMS6 | 138607 | 132501 | 95.59 | 227.4 | 53.275 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Tian, J.; Cidan, Y.; Wang, H.; Li, K.; Basang, W. Influence of Varied Environment Conditions on the Gut Microbiota of Yaks. Animals 2024, 14, 1570. https://doi.org/10.3390/ani14111570
Zhu Y, Tian J, Cidan Y, Wang H, Li K, Basang W. Influence of Varied Environment Conditions on the Gut Microbiota of Yaks. Animals. 2024; 14(11):1570. https://doi.org/10.3390/ani14111570
Chicago/Turabian StyleZhu, Yanbin, Jiayi Tian, Yangji Cidan, Hongzhuang Wang, Kun Li, and Wangdui Basang. 2024. "Influence of Varied Environment Conditions on the Gut Microbiota of Yaks" Animals 14, no. 11: 1570. https://doi.org/10.3390/ani14111570
APA StyleZhu, Y., Tian, J., Cidan, Y., Wang, H., Li, K., & Basang, W. (2024). Influence of Varied Environment Conditions on the Gut Microbiota of Yaks. Animals, 14(11), 1570. https://doi.org/10.3390/ani14111570